GFP Fusion Proteins: A Complete Guide to Visualizing and Analyzing Protein-Protein Interactions in Living Cells
This comprehensive guide explores the principles and applications of GFP (Green Fluorescent Protein) fusion techniques for monitoring protein-protein interactions (PPIs) in real-time within living cells.
GFP Fusion Proteins: A Complete Guide to Visualizing and Analyzing Protein-Protein Interactions in Living Cells
Abstract
This comprehensive guide explores the principles and applications of GFP (Green Fluorescent Protein) fusion techniques for monitoring protein-protein interactions (PPIs) in real-time within living cells. It begins by establishing the foundational science of fluorescent proteins and their adaptation for FRET/BRET-based interaction assays. The article provides a detailed methodological walkthrough for designing and implementing GFP-based PPI sensors, including BiFC, FRET, and modern split-GFP systems. It addresses common experimental challenges, offering troubleshooting strategies for issues like false positives, background fluorescence, and expression artifacts. Finally, the guide critically evaluates the validation requirements for these techniques, comparing them to alternative methods like yeast two-hybrid and co-immunoprecipitation. Designed for researchers and drug discovery professionals, this resource aims to equip scientists with the knowledge to reliably apply and interpret GFP fusion assays in biomedical research.
What are GFP Fusion Proteins? The Science Behind Visualizing PPIs in Live Cells
Application Notes: GFP-Based Protein-Protein Interaction Assays
The discovery of Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria and its subsequent engineering has revolutionized live-cell imaging. Within the context of a thesis on GFP fusions for monitoring protein-protein interactions (PPIs), these tools enable real-time, spatial, and quantitative analysis of dynamic molecular events in their native cellular environment. Key applications include fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation (BiFC), and fluorescence correlation spectroscopy (FCS).
Table 1: Comparison of Common Fluorescent Proteins for PPI Studies
| Fluorescent Protein | Excitation Max (nm) | Emission Max (nm) | Brightness (Relative to EGFP) | Maturation Half-time (37°C) | Oligomeric State | Primary PPI Application |
|---|---|---|---|---|---|---|
| EGFP | 488 | 507 | 1.0 | ~30 min | Monomeric | Standard fusion tag |
| mCherry | 587 | 610 | 0.47 | ~15 min | Monomeric | FRET acceptor, co-localization |
| Cerulean (CFP variant) | 433 | 475 | 0.49 | ~45 min | Monomeric | FRET donor |
| Venus (YFP variant) | 515 | 528 | 1.56 | ~15 min | Monomeric | FRET acceptor, BiFC |
| mNeonGreen | 506 | 517 | 2.4 | ~10 min | Monomeric | High-signal fusion tag |
| TagRFP-T | 555 | 584 | 0.81 | ~1.4 hr | Monomeric | FRET, photostable acceptor |
Table 2: Performance Metrics of PPI Assay Techniques Using GFP Variants
| Assay Technique | Typical GFP Pair Used | Approx. Detection Range (nm) | Key Measurable Output | Advantages | Limitations |
|---|---|---|---|---|---|
| FRET (Acceptor Photobleaching) | CFP/YFP (e.g., Cerulean/Venus) | 1-10 nm | Donor dequenching efficiency | Direct interaction proof, quantitative | Photobleaching irreversible, slow |
| FRET (Sensitized Emission) | GFP/mCherry | 1-10 nm | FRET efficiency (E%) | Ratiometric, live-cell kinetics | Requires careful correction |
| BiFC | Split-Venus (YN/YC) | N/A (irreversible) | Reconstituted fluorescence signal | High signal-to-noise, captures weak/transient | Irreversible complementation |
| Fluorescence Cross-Correlation Spectroscopy (FCCS) | EGFP/mCherry | Diffraction-limited volume | Diffusion coefficient, binding coefficient | Quantifies binding stoichiometry & dynamics | Requires specialized microscopy |
Experimental Protocols
Protocol 1: FRET by Sensitized Emission for Live-Cell PPI Analysis
Objective: To quantify the interaction between two proteins of interest (Protein A and Protein B) using Cerulean (CFP donor) and Venus (YFP acceptor) fusions.
Research Reagent Solutions Toolkit:
| Reagent/Material | Function/Explanation |
|---|---|
| pCerulean-N1 Vector | Donor FP plasmid backbone for N-terminal fusion. |
| pVenus-C1 Vector | Acceptor FP plasmid backbone for C-terminal fusion. |
| Lipofectamine 3000 | Transfection reagent for mammalian cell delivery. |
| Dulbecco’s Modified Eagle Medium (DMEM), phenol-red free | Imaging-optimized cell culture medium. |
| CO2-independent Imaging Medium | Maintains pH during microscopy. |
| HEK 293T Cells | Easily transfectable mammalian cell line. |
| Poly-D-Lysine Coated Coverslips | Enhances cell adherence for imaging. |
| Confocal Microscope with Spectral Detectors | Equipped with 405nm and 514nm lasers for CFP/YFP excitation. |
Methodology:
- Construct Preparation: Subclone cDNA for Protein A into pCerulean-N1 and Protein B into pVenus-C1. Verify sequences.
- Cell Seeding & Transfection: Seed HEK 293T cells on coated coverslips in a 24-well plate. At 60-70% confluency, co-transfect with 0.5 µg of each plasmid using Lipofectamine 3000 per manufacturer’s protocol.
- Expression: Incubate cells for 24-48 hours at 37°C, 5% CO2.
- Microscopy Setup: Mount coverslip in imaging chamber with CO2-independent medium. Use a 63x oil immersion objective.
- Donor Channel: Excite at 405nm, collect emission at 470±20nm.
- Acceptor Channel: Excite at 514nm, collect emission at 535±20nm.
- FRET Channel: Excite at 405nm, collect emission at 535±20nm.
- Image Acquisition & Correction: Acquire images for all three channels. Include cells expressing Cerulean-only and Venus-only for spectral bleed-through (SBT) correction.
- FRET Efficiency Calculation: Use the corrected FRET (cFRET) formula:
cFRET = FRETraw - (a * Donor) - (b * Acceptor), whereaandbare SBT coefficients. Calculate FRET efficiency:E = cFRET / (cFRET + G * Donor).Gis an instrument-specific calibration factor.
Protocol 2: Bimolecular Fluorescence Complementation (BiFC) Assay
Objective: To visualize and confirm a protein-protein interaction via reconstitution of a split Venus fluorescent protein.
Research Reagent Solutions Toolkit:
| Reagent/Material | Function/Explanation |
|---|---|
| Split Venus Vectors (pYN155, pYC155) | Plasmids encoding Venus N-terminal (1-155) and C-terminal (156-238) fragments. |
| Positive Control Plasmids (e.g., Fos-YN/Jun-YC) | Validated interacting pair for assay validation. |
| Negative Control Plasmid (e.g., unfused YN/YC) | Controls for spontaneous complementation. |
| Hoechst 33342 Stain | Nuclear counterstain for cell localization. |
| Paraformaldehyde (4%) | Fixative for endpoint analysis. |
| Mounting Medium (Antifade) | Preserves fluorescence for imaging. |
Methodology:
- Construct Generation: Fuse Protein A to the N-terminal fragment (YN) and Protein B to the C-terminal fragment (YC).
- Cell Transfection: Seed appropriate cells (e.g., HeLa) in 6-well plates. Co-transfect pairs: Experimental (Protein A-YN + Protein B-YC), Positive Control, and Negative Control.
- Incubation: Incubate for 24-36 hours. The complementation and fluorophore maturation require time.
- Fixation (Optional): Wash cells with PBS, fix with 4% PFA for 15 min, and mount with antifade medium containing Hoechst.
- Imaging: Use a standard epifluorescence or confocal microscope with a YFP filter set (excitation ~500nm, emission ~535nm). Detect signal only if Proteins A and B interact and bring the split Venus fragments into proximity.
- Analysis: Qualitatively assess interaction by fluorescence localization and intensity compared to controls. Quantification can be performed via fluorescence intensity measurement in defined cellular regions.
Visualization Diagrams
Diagram Title: GFP Development and PPI Assay Pathways
Diagram Title: FRET Sensitized Emission Protocol Workflow
Diagram Title: BiFC Assay Mechanism for Detecting PPIs
Application Notes
Within the broader thesis investigating GFP fusions for monitoring protein-protein interactions, tagging proteins with GFP is the foundational technique enabling the direct visualization of these dynamics in living systems. The core principle is the creation of a functional fusion protein, where the gene for Green Fluorescent Protein (or its variants) is genetically linked to the gene of the target protein. This chimera is expressed in cells, resulting in a target protein that is intrinsically fluorescent. When illuminated with specific wavelengths of light (typically blue light), GFP emits green fluorescence, allowing researchers to track the localization, movement, and interactions of the tagged protein in real time without fixing or killing the cells. This section details the quantitative parameters and experimental workflows central to implementing this principle.
Table 1: Key Quantitative Parameters for GFP-based Live-Cell Imaging
| Parameter | Typical Range / Value | Significance for Protein-Protein Interaction Studies |
|---|---|---|
| Excitation/Emission Max (eGFP) | Ex: 488 nm / Em: 507 nm | Standard filter sets allow specific detection with minimal background. |
| Maturation Time (eGFP, 37°C) | ~30-90 minutes | Critical for pulse-chase or rapid turnover experiments; delay between synthesis and fluorescence. |
| Photostability (t1/2, under imaging) | Varies; ~50-100s for continuous illumination | Limits duration of continuous imaging; use lower intensity or opt for more photostable variants (e.g., mNeonGreen). |
| pKa of Chromophore | ~6.0 | Fluorescence is quenched in acidic environments (e.g., lysosomes), affecting quantification in certain compartments. |
| Detection Limit (Molecules/µm²) | ~50-100 molecules/µm² | Defines the minimum expression level required for reliable detection of low-abundance proteins. |
| FRET Efficiency (for Interaction) | 5-35% | Range of energy transfer efficiency measurable when using GFP variants as FRET pairs (e.g., CFP/YFP). |
Protocols
Protocol 1: Generation of a Mammalian Expression Plasmid for N-Terminal GFP Fusion
Objective: To clone the coding sequence of your protein of interest (POI) downstream of the GFP sequence in a mammalian expression vector for transient or stable expression.
Materials:
- Template: cDNA for POI.
- Vector: e.g., pEGFP-N1 or pEGFP-C1 (Takara Bio).
- Enzymes: Restriction enzymes, T4 DNA Ligase, high-fidelity DNA polymerase.
- Cells: Competent E. coli (DH5α).
- Reagents: LB broth/agar with appropriate antibiotic (Kanamycin), gel extraction kit, plasmid miniprep kit.
Method:
- Amplify POI Gene: Design primers to amplify the POI open reading frame (ORF). Forward primer: Include a restriction site (e.g., AgeI) compatible with the vector, followed by a flexible linker sequence (e.g., GGT GGC GGC TCC GG). Reverse primer: Include a stop codon and a different restriction site (e.g., NotI).
- Digestion: Purify PCR product. Digest both the purified PCR product and the pEGFP-C1 vector with AgeI and NotI.
- Ligation: Purify digested fragments. Set up a ligation reaction with a 3:1 (insert:vector) molar ratio using T4 DNA Ligase. Incubate at 16°C for 1 hour.
- Transformation & Screening: Transform ligation mix into competent E. coli. Plate on LB-Kanamycin plates. Pick colonies, culture, and isolate plasmid DNA.
- Verification: Confirm correct insertion and reading frame by restriction digest and Sanger sequencing using a vector-specific primer upstream of the MCS.
Protocol 2: Live-Cell Confocal Imaging of GFP-Tagged Protein Dynamics
Objective: To image the subcellular localization and dynamics of a GFP-tagged protein in living mammalian cells.
Materials:
- Cells: HeLa or HEK293T cells transfected with the GFP-POI construct.
- Imaging Dish: Glass-bottom 35 mm culture dish (e.g., µ-Dish, ibidi).
- Microscope: Confocal microscope with a 488 nm laser line, 40x or 63x oil immersion objective, and environmental chamber (37°C, 5% CO₂).
- Media: Live-cell imaging medium (FluoroBrite DMEM, Thermo Fisher) supplemented with 10% FBS and 25 mM HEPES.
Method:
- Cell Preparation: Plate cells in the glass-bottom dish 24 hours prior. Transfect with the GFP-POI construct using your preferred method. Perform imaging 24-48 hours post-transfection.
- Microscope Setup: Pre-warm the environmental chamber to 37°C. Replace culture medium with pre-warmed live-cell imaging medium.
- Acquisition Settings:
- Use the 488 nm laser at low power (1-10%) to minimize phototoxicity.
- Set emission detection window to 500-550 nm.
- Set pinhole to 1 Airy unit for optimal optical sectioning.
- Use a high-speed, low-noise detector (e.g., GaAsP PMT).
- Adjust gain and offset to utilize the full dynamic range without saturation.
- Time-Lapse Acquisition: Define imaging region(s). Set time interval (e.g., every 10-30 seconds for fast dynamics, 5-15 minutes for slower processes). Set total duration. Begin acquisition.
- Analysis: Use image analysis software (e.g., Fiji/ImageJ) to generate kymographs, measure fluorescence intensity over time in regions of interest (ROIs), or track particles.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in GFP Live-Cell Imaging |
|---|---|
| pEGFP-C1/N1 Vectors | Standard mammalian expression vectors for creating C- or N-terminal fusions; contain CMV promoter for strong expression. |
| FluoroBrite DMEM | Low-fluorescence, phenol red-free medium that drastically reduces background autofluorescence during live imaging. |
| Glass-bottom Culture Dishes | Provide optimal optical clarity for high-resolution microscopy while allowing cell growth. |
| FuGENE HD Transfection Reagent | A low-toxicity, high-efficiency reagent for delivering plasmid DNA into a wide range of mammalian cell lines. |
| Hoechst 33342 (or SiR-DNA) | Cell-permeable nuclear counterstain (blue or far-red) for visualizing nuclei without significant spectral overlap with GFP. |
| Vectashield Antifade Mountant (Live) | Reagent to reduce photobleaching during prolonged live imaging sessions. |
Visualizations
GFP Fusion Protein Creation & Imaging Workflow
Principle of GFP Excitation and Emission
GFP Fusion Workflow for Interaction Studies
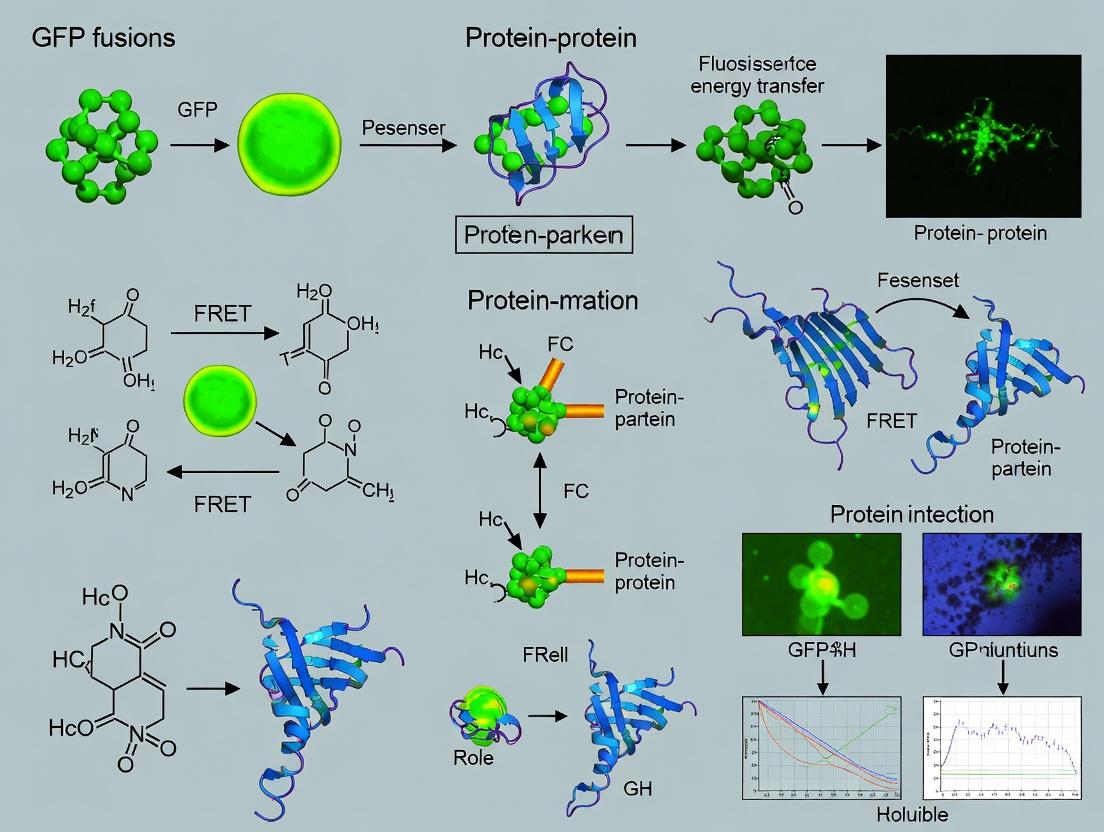
Within the broader thesis on GFP-based technologies for monitoring protein-protein interactions (PPIs), Förster Resonance Energy Transfer (FRET), Bimolecular Fluorescence Complementation (BiFC), and Split-GFP represent cornerstone methodologies. Each assay provides unique insights into the dynamics, localization, and specificity of PPIs, driving discovery in cell biology and drug development.
Förster Resonance Energy Transfer (FRET)
FRET measures proximity (1-10 nm) between two fluorophores, a donor and an acceptor. Upon donor excitation, energy transfer to the acceptor occurs only if they are in extremely close proximity, leading to acceptor emission. When fused to candidate interacting proteins, FRET efficiency serves as a quantitative molecular ruler for interaction.
Application Notes: Ideal for studying real-time interaction kinetics and conformational changes in living cells. Commonly used donor-acceptor pairs are CFP-YFP (e.g., CyPet-YPet) or the modern green-red pair mNeonGreen-mRuby2. Sensitive to precise spectral calibration and prone to false positives from overexpression or bleed-through.
Quantitative Comparison of Common FRET Pairs:
| FRET Pair (Donor->Acceptor) | Förster Radius (R₀ in nm) | Dynamic Range (ΔF/F₀%) | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| CFP (CyPet) -> YFP (YPet) | 4.9 - 5.2 | 15-30% | Genetically encoded; well-characterized | Significant spectral bleed-through |
| GFP -> mCherry | ~4.5 | 10-20% | Bright, photostable acceptor | Smaller R₀, lower FRET efficiency |
| mNeonGreen -> mRuby2 | ~5.6 | 25-40% | High brightness, excellent separation | Requires optimized filter sets |
| BFP -> GFP | ~4.0 | 5-15% | Minimal cross-excitation | Low donor brightness, photobleaching |
Protocol: Acceptor Photobleaching FRET Assay (Confocal Microscopy) Objective: To confirm proximity by measuring donor de-quenching after acceptor destruction.
- Construct Preparation: Clone proteins of interest (POI-A, POI-B) into vectors encoding donor (e.g., CFP) and acceptor (e.g., YFP), respectively.
- Cell Transfection: Co-transfect constructs into HEK293T cells using a 1:3 DNA:PEI ratio. Culture for 24-48 hours.
- Image Acquisition: Using a 63x oil objective, capture pre-bleach donor (CFP ex: 458nm, em: 470-500nm) and acceptor (YFP ex: 514nm, em: 525-550nm) channels.
- Acceptor Photobleaching: Define a region of interest (ROI) containing the complex. Bleach the acceptor using 514nm laser at 100% power for 5-15 iterations.
- Post-Bleach Imaging: Re-image the donor channel under identical settings.
- Data Analysis: Calculate FRET efficiency: E = 1 – (Donor_pre / Donor_post). A significant increase in donor fluorescence post-bleach indicates positive FRET.
Bimolecular Fluorescence Complementation (BiFC)
BiFC uses split fragments of a fluorescent protein (e.g., YFP) fused to putative interacting partners. Interaction brings the fragments together, facilitating reconstitution and fluorescence. This is an irreversible complementation assay.
Application Notes: Excellent for detecting weak or transient interactions, determining subcellular localization of PPIs, and visualizing multiple complexes simultaneously using different split FP colors (multicolor BiFC). Major drawback: complementation is irreversible and can trap complexes, potentially creating artifacts.
Protocol: Standard BiFC Assay for Nuclear Interactions
- Vector System: Use split Venus YFP (VN173, VC155) or split mCherry vectors. Fuse POI-X to N-terminal fragment (VN) and POI-Y to C-terminal fragment (VC).
- Controls: Include pairs of known interactors (positive control) and non-interactors (e.g., VC fused to empty tag).
- Transfection: Seed HeLa cells in 8-well chamber slides. Transfect with 100ng each BiFC plasmid using lipofection reagent. Incubate 24-36h.
- Imaging & Analysis: Image using a standard YFP filter set. Score fluorescence intensity and localization. Quantify mean fluorescence per cell using ImageJ. Note: Include a nuclear marker (e.g., DAPI) for co-localization verification.
Split-GFP
Split-GFP employs a small, non-fluorescent GFP fragment (GFP11, ~16 aa) tagged to a POI, which complements with a larger GFP fragment (GFP1-10) expressed in the host cell or in vitro. Fluorescence only upon binding.
Application Notes: Highly specific with low background due to minimal spontaneous complementation. Versatile for detecting interactions, protein trafficking, and generating stable cell lines where GFP1-10 is constitutively expressed. The GFP11 tag is minimally invasive.
Protocol: Detecting Membrane Protein Interactions with Split-GFP
- Stable Cell Line Generation: Create a HEK293 cell line stably expressing the GFP1-10 detector fragment using lentiviral transduction and antibiotic selection.
- Sensor Introduction: Transfect these cells with plasmid(s) encoding your POI fused to the GFP11 tag.
- Live-Cell Monitoring: Image 24-72 hours post-transfection using a standard GFP filter set. Fluorescence indicates successful delivery and complementation.
- Quantification (Flow Cytometry): Harvest cells, resuspend in PBS, and analyze on a flow cytometer (ex: 488nm, em: 510/20nm). The percentage of GFP-positive cells and median fluorescence intensity quantify interaction or expression levels.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent / Material | Function & Application |
|---|---|
| PEI Max (Polyethylenimine) | High-efficiency, low-cost polymeric transfection reagent for plasmid DNA delivery. |
| Fluorescent Protein Vectors | Donor/Acceptor (FRET), Split-FP (BiFC, Split-GFP) plasmids from addgene or commercial sources. |
| Cell Lines (HEK293T, HeLa) | Robust, easily transfected mammalian lines for proof-of-concept interaction studies. |
| Antibiotic Selection Markers | For generating stable cell lines (e.g., with GFP1-10), using puromycin, blasticidin, etc. |
| Live-Cell Imaging Medium | Phenol-red free medium with HEPES buffer for maintaining pH during microscopy. |
| Mounting Medium with DAPI | For fixing cells and staining nuclei in endpoint BiFC/FRET experiments. |
| Spectral Detector Confocal | Microscope capable of lambda scanning or optimized filter sets for FRET quantification. |
Visualization Diagrams
Diagram 1: FRET Energy Transfer Mechanism
Diagram 2: BiFC & Split-GFP Workflow Comparison
Diagram 3: Experimental Decision Pathway
Within the broader thesis on the application of GFP fusions for monitoring protein-protein interactions (PPIs), understanding the rationale for live-cell analysis is fundamental. Traditional biochemical methods, while foundational, provide a static and often disrupted snapshot of interactions. This document outlines the critical advantages of live-cell PPI monitoring and provides detailed application notes and protocols centered on Förster Resonance Energy Transfer (FRET) using GFP variants, the current gold standard for quantitative, dynamic PPI analysis in living cells.
Comparative Advantages: Live-Cell vs. Traditional Methods
Table 1: Comparison of PPI Analysis Methods
| Feature | Traditional Biochemical Methods (Co-IP, Y2H) | Live-Cell Imaging (e.g., FRET/BRET) |
|---|---|---|
| Cellular Context | Lysed cells, no context. | Intact, living cells. |
| Temporal Resolution | Endpoint measurement. | Real-time, kinetic data (seconds-minutes). |
| Spatial Information | None. | Subcellular localization of interaction. |
| Interaction Dynamics | Static snapshot. | Dynamic assembly/disassembly in response to stimuli. |
| Physiological Relevance | Non-physiological buffers, potential artifacts. | Native environment, correct pH, ions, and organelle contacts. |
| Throughput | Low to moderate. | Moderate to high (in 96/384-well plates). |
| Key Limitation | Cannot detect transient or weak interactions. | Technical sensitivity, donor-acceptor ratio, photobleaching. |
Protocol 1: Live-Cell FRET by Acceptor Photobleaching (Quantitative)
This protocol measures the increase in donor fluorescence after selective bleaching of the acceptor, providing a robust quantitative FRET efficiency (E) calculation.
Materials & Reagent Solutions
- Plasmid Vectors: pEGFP-N1 (Donor), pmCherry-C1 (Acceptor). Ensure in-frame fusion to proteins of interest (POIs).
- Cell Line: HEK293T or HeLa, cultured in appropriate medium (DMEM + 10% FBS).
- Transfection Reagent: Polyethylenimine (PEI) or Lipofectamine 3000.
- Imaging Medium: FluoroBrite DMEM or CO₂-independent medium.
- Microscope: Confocal microscope with 405, 488, and 561 nm lasers, and controllable region of interest (ROI) bleaching capability.
Procedure:
- Transfection: Co-transfect cells with donor (GFP-POI) and acceptor (mCherry-POI) plasmids at a 1:1 molar ratio on glass-bottom dishes. Include controls: donor alone, acceptor alone.
- Expression: Incubate for 24-48h to achieve moderate expression.
- Image Acquisition: Select cells expressing both fluorophores at comparable levels.
- Acquire pre-bleach images: Donor channel (Ex 488nm / Em 500-550nm) and Acceptor channel (Ex 561nm / Em 570-620nm).
- Acceptor Photobleaching: Define an ROI containing the interaction site. Bleach acceptor with high-power 561 nm laser (70-100% power, 5-20 iterations).
- Post-bleach Acquisition: Re-acquire donor and acceptor channel images under identical pre-bleach settings.
- Quantification:
- Measure mean donor fluorescence intensity in the ROI before (Dpre) and after (Dpost) bleaching.
- Calculate FRET Efficiency: E = (Dpost - Dpre) / D_post.
- Verify >70% acceptor bleach (loss of mCherry signal).
Diagram 1: FRET Acceptor Photobleaching Workflow
Protocol 2: Kinetic BRET Assay for GPCR-Arrestin Interaction (High-Throughput)
Bioluminescence Resonance Energy Transfer (BRET) is ideal for kinetic, plate-reader based studies, such as monitoring GPCR signaling.
Materials & Reagent Solutions
- Plasmid Vectors: GPCR fused to NanoLuc luciferase (NLuc), β-arrestin fused to HaloTag.
- Cell Line: HEK293 expressing relevant G-proteins.
- Substrate: Furimazine (commercial NLuc substrate).
- Ligand: Specific agonist/antagonist for the GPCR.
- HaloTag Ligand: HaloTag 618 Ligand (fluorescent acceptor).
- Instrument: Plate reader capable of sequential luminescence/filtered fluorescence detection.
Procedure:
- Cell Preparation: Seed cells in a white 96-well plate. Co-transfect with NLuc-GPCR and HaloTag-arrestin.
- Labeling: 24h post-transfection, add HaloTag 618 Ligand to medium (final ~100 nM). Incubate 15-30 min, wash.
- Equilibration: Add fresh imaging medium containing Furimazine.
- Kinetic Measurement: Place plate in reader. Establish a baseline luminescence reading (475nm filter for donor).
- Inject agonist via injector port.
- Immediately start dual-emission kinetic recording: Donor (475±20nm) and BRET (610±10nm) every 10-60 seconds for 30+ minutes.
- Data Analysis:
- Calculate BRET ratio = (Acceptor emission) / (Donor emission).
- Plot BRET ratio vs. time. Normalize to baseline.
- Derive kinetic parameters (t½, max response).
Diagram 2: GPCR-Arrestin BRET Signaling Pathway & Readout
The Scientist's Toolkit: Key Reagents for Live-Cell PPI
Table 2: Essential Research Reagents for Live-Cell PPI Studies
| Reagent Category | Specific Example | Function in Experiment |
|---|---|---|
| Fluorescent Protein Donor | GFP, mTurquoise2, NanoLuc | Genetically encoded tag; serves as FRET/BRET energy donor. |
| Fluorescent Protein Acceptor | mCherry, YFP, HaloTag-618 Ligand | Energy acceptor; emission indicates proximity (<10nm). |
| Bioluminescent Donor | NanoLuc Luciferase | Superior BRET donor; high brightness, stability. |
| Substrate | Furimazine | NanoLuc substrate; produces luminescence for BRET. |
| Live-Cell Dyes | HaloTag, SNAP-tag Ligands | Covalently label self-labeling tags for flexible acceptor introduction. |
| Validated Biosensor | Erk Kinase Translocation Reporter (EKAR) | Pre-optimized FRET biosensor for specific pathway activity. |
| Transfection Reagent | Polyethylenimine (PEI), Lipofectamine 3000 | Introduces plasmid DNA encoding fusion proteins into cells. |
| Opti-MEM Reduced Serum Medium | Gibco Opti-MEM | Low-serum medium for complex formation during transfection. |
Step-by-Step Protocols: Designing and Implementing GFP-Based PPI Assays
Application Notes
Within a thesis investigating protein-protein interactions (PPIs) using GFP fusions, the design of the fusion construct is a critical determinant of experimental success. The choice between N-terminal and C-terminal fusion, coupled with the selection of an appropriate linker, directly impacts the solubility, stability, localization, and functional integrity of both the protein of interest (POI) and the fluorescent reporter.
Key Considerations:
- N-terminal Fusions place GFP at the start of the POI. This can be advantageous if the C-terminus of the POI is involved in interactions, localization, or function. However, it may interfere with N-terminal signal peptides or modification sites.
- C-terminal Fusions attach GFP to the end of the POI. This strategy protects N-terminal features but can disrupt C-terminal functional elements such as prenylation sites or endoplasmic reticulum retention signals.
- Linker Design is essential to provide spatial separation and flexibility, reducing steric hindrance between domains. Optimal linkers prevent misfolding and maintain the independent folding and function of both moieties.
The optimal configuration must be determined empirically for each POI. A systematic approach, testing both orientations with a selection of linkers, is recommended to identify the construct that best reports on the native behavior and interactions of the POI.
Experimental Protocols
Protocol 1: Modular Golden Gate Assembly for Testing Fusion Variants
This protocol enables the high-throughput cloning of a single POI with GFP in multiple configurations (N/C-terminal, different linkers) using Golden Gate Assembly.
Materials: Destination vector with acceptor site (e.g., BsaI sites), Entry vectors for: GFP (no start/stop), N-terminal linker library, C-terminal linker library, PCR-amplified POI (with overhangs compatible with linker fragments), T4 DNA Ligase, BsaI-HFv2 restriction enzyme, ThermoCycler.
Procedure:
- Prepare Fragments: Digest entry vectors and purify the fragments: POI, GFP, chosen N-linker(s), chosen C-linker(s). Alternatively, use PCR to generate fragments with appropriate 4-bp overhangs.
- Assembly Reaction: Set up a 20 µL reaction containing:
- 50 ng destination vector
- Equimolar amounts of POI, GFP, and linker fragments (~20 fmol each)
- 1 µL T4 DNA Ligase
- 1 µL BsaI-HFv2
- 1X T4 DNA Ligase Buffer
- Cycled Assembly: Place in a thermocycler: (37°C for 2 min, 16°C for 5 min) x 25 cycles, followed by 50°C for 5 min, 80°C for 10 min.
- Transformation: Transform 2 µL of the reaction into competent E. coli, plate on selective media, and screen colonies by colony PCR or sequencing.
Protocol 2: Confocal Microscopy Validation of Fusion Protein Localization
Materials: Mammalian cells (e.g., HEK293), transfection reagent, constructed GFP fusion plasmids, live-cell imaging chamber, confocal microscope.
Procedure:
- Transfection: Seed cells on glass-bottom dishes. At 60-80% confluency, transfect with each GFP fusion construct using a standard protocol.
- Expression: Incubate for 18-48 hours to allow expression.
- Imaging: Replace medium with live-cell imaging buffer. Using a confocal microscope with a 488 nm laser and appropriate emission filter:
- Acquire GFP fluorescence images.
- Acquire differential interference contrast (DIC) or phase-contrast images.
- If available, co-stain with organelle-specific dyes (e.g., MitoTracker, ER-Tracker) for co-localization analysis.
- Analysis: Compare the localization pattern of each GFP fusion (N vs. C, different linkers) to the expected, well-characterized localization of the untagged POI.
Data Presentation
Table 1: Comparative Analysis of Fusion Site Outcomes for Model Proteins
| Protein (Localization) | Fusion Site | Linker Used (Length) | Result: Correct Localization? | Result: Interaction Reported? | Notes |
|---|---|---|---|---|---|
| p53 (Nuclear) | C-terminal | (GGS)₅ (15 aa) | Yes | Yes (with MDM2) | N-terminal fusion showed cytoplasmic aggregation. |
| K-Ras (Plasma Membrane) | C-terminal | (GGS)₃ (9 aa) | No | N/A | C-terminal fusion disrupted farnesylation. N-terminal fusion required. |
| Cytochrome c (Mitochondrial) | N-terminal | (EAAAK)₃ (15 aa) | Yes | Yes (in apoptosis assays) | Rigid helical linker helped maintain independent folding. |
| Calreticulin (ER Luminal) | C-terminal | Flexible (GGGGS)₄ (20 aa) | Yes | Partial | Long flexible linker needed to span ER membrane. Signal peptide must remain N-terminal. |
Table 2: Properties of Common Linker Types
| Linker Type | Sequence Example | Length (AA) | Predicted Flexibility | Common Application |
|---|---|---|---|---|
| Flexible | (GGGGS)ₙ | 5n | High | General use, separating independent domains. |
| Rigid | (EAAAK)ₙ | 5n | Low | Maintaining distance, preventing unwanted interaction. |
| Cleavable | LEVLFQ/GP (TEV site) | 7 | Variable | Removing tag after purification in vitro. |
| β-sheet forming | (XP)ₙ | Variable | Medium | To fix orientation, often in scFv fragments. |
Visualizations
Title: Decision Workflow for GFP Fusion Construct Design
Title: Monitoring MAPK Pathway Interactions with GFP Fusions
The Scientist's Toolkit
Table 3: Essential Reagents for GFP Fusion Construct Research
| Reagent / Solution | Function / Purpose |
|---|---|
| Cloning Kit (Golden Gate or Gibson) | Enables modular, seamless assembly of multiple DNA fragments (POI, GFP, linkers). |
| Flexible Linker Plasmid Library | A set of vectors containing different (GGGGS)ₙ repeats for easy insertion. |
| Rigid Linker Oligonucleotides | Pre-designed oligos for synthesizing helical (EAAAK)ₙ linkers via PCR. |
| Live-Cell Imaging Medium | Phenol-red free, HEPES-buffered medium to maintain pH and health during microscopy. |
| Organelle-Specific Dyes (e.g., MitoTracker Deep Red) | For co-localization studies to validate correct subcellular targeting of the fusion. |
| Protease Inhibitor Cocktail (EDTA-free) | Critical during protein extraction for downstream biochemical validation (e.g., Co-IP). |
| Anti-GFP Nanobody Agarose | For standardized immunoprecipitation of any GFP fusion protein to test interactions. |
| TEV Protease | For cleaving and removing the GFP tag in vitro after purification for functional assays. |
Vector Selection and Cloning Strategies for GFP Fusion Proteins
Within the broader thesis investigating GFP fusions for monitoring protein-protein interactions (PPIs), the initial and critical step is the construction of a functional fusion protein. The selection of an appropriate expression vector and the implementation of a robust cloning strategy directly determine the success of downstream interaction assays. This application note details current best practices, focusing on generating fusion constructs that preserve the native localization, stability, and interaction capabilities of the protein of interest (POI).
Vector Selection Criteria
The choice of vector is paramount. Key considerations are summarized in the table below.
Table 1: Key Criteria for Vector Selection for GFP Fusion Proteins
| Criterion | Options | Considerations for PPI Research |
|---|---|---|
| Fusion Orientation | N-terminal GFP, C-terminal GFP | May affect POI folding, function, or interaction interfaces. Empirical testing is often required. |
| Promoter | Constitutive (CMV, EF1α), Inducible (Tet-On/Off), Tissue/Cell-specific | Must drive expression appropriate for the model system and avoid overexpression artifacts that perturb PPIs. |
| Selection Marker | Antibiotic (Ampicillin, Kanamycin), Auxotrophic, Fluorescent | Dictates selection in bacteria and mammalian cells. Dual selection markers aid in stable cell line generation. |
| Tagging System | Single GFP, Tandem Tags (e.g., SFB, FLAG-HA), Split GFP | Tandem tags facilitate sequential purification for PPI validation. Split GFP can be used for bimolecular complementation assays. |
| Cloning Method | Restriction Enzyme, Gateway, In-Fusion, Gibson Assembly | Impacts speed, flexibility, and the ability to create multiple constructs in parallel. |
| Copy Number & Origin | High (pUC), Low (pSC101), Mammalian (Episomal) | Bacterial copy number affects plasmid yield and stability. Episomal mammalian origins simplify genomic integration studies. |
Core Cloning Strategies
Restriction Enzyme-Based Cloning
A traditional but reliable method for inserting a POI into a GFP vector.
Protocol: Standard Restriction Enzyme Cloning for GFP Fusions
- Design: Analyze the multiple cloning site (MCS) of the destination GFP vector. Select restriction enzymes that are unique to the MCS and absent from the POI coding sequence.
- PCR Amplification: Amplify the POI cDNA using high-fidelity DNA polymerase with gene-specific primers containing the chosen restriction sites and a 4-6 bp 5' overhang.
- Digestion: Digest both the purified PCR product and the destination vector with the selected restriction enzymes. Use alkaline phosphatase (e.g., CIP, SAP) to dephosphorylate the vector ends to prevent self-ligation.
- Purification: Gel-purify the digested POI insert and linearized vector.
- Ligation: Mix insert and vector at a molar ratio (typically 3:1 to 5:1 insert:vector) with T4 DNA Ligase. Incubate at 16°C for 1-2 hours or overnight.
- Transformation: Transform the ligation mix into competent E. coli, plate on selective antibiotic media, and incubate overnight.
- Screening: Screen colonies by colony PCR or restriction digest of miniprep DNA. Confirm the sequence of the final GFP::POI or POI::GFP construct by Sanger sequencing.
Gateway Recombination Cloning
An efficient, site-specific recombination system ideal for high-throughput transfer of a POI into multiple GFP-fusion destination vectors.
Protocol: Gateway Cloning for GFP Fusion Construction
- Entry Clone Creation: Clone the POI into a donor vector (e.g., pDONR) via BP recombination between attB sites (on primers) and attP sites (on vector). This generates an entry clone with attL-flanked POI.
- LR Reaction: Mix the entry clone with the desired GFP-fusion destination vector (containing attR sites and a lethal gene like ccdB). Add LR Clonase II enzyme mix.
- Recombination: The LR reaction (typically 1 hour at 25°C) transfers the POI from the entry clone into the destination vector, replacing the ccdB gene and creating the final expression clone (with attB sites).
- Transformation & Selection: Transform the reaction into competent E. coli. Selection on the appropriate antibiotic (determined by the destination vector) ensures growth of only cells with the recombined expression clone.
- Verification: Confirm clone size by analytical digest and validate the fusion junction by sequencing.
Critical Considerations for PPI Studies
- Linker Design: A flexible peptide linker (e.g., (GGGGS)n) between GFP and the POI is often essential to minimize steric hindrance and allow proper folding of both moieties.
- Subcellular Localization: Verify that the GFP fusion recapitulates the known localization of the native POI. Mislocalization suggests folding or targeting issues.
- Functional Validation: The GFP fusion should retain the biological activity of the POI. Perform a rescue experiment in a POI-deficient cell line or a known functional assay.
- Controls: Always include unfused GFP and untagged POI controls to distinguish artifacts from genuine interactions in co-localization or co-immunoprecipitation experiments.
Visualization of Workflows
Title: Gateway Cloning for GFP Fusion Construction
Title: From GFP Fusion Construct to PPI Assay
The Scientist's Toolkit
Table 2: Essential Research Reagents & Materials
| Reagent / Material | Function & Importance |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | Ensures accurate PCR amplification of the POI with minimal errors for reliable fusion sequences. |
| Gateway BP & LR Clonase II Enzyme Mix | Enables efficient, directional recombination for Gateway cloning workflows. |
| Competent E. coli Cells (Cloning & Expression Strains) | For plasmid propagation and, if using bacterial expression, for recombinant protein production. |
| Mammalian Cell Line (e.g., HEK293T, HeLa) | A standard, easily transfectable system for expressing GFP fusions and performing initial PPI assays. |
| Lipid-Based Transfection Reagent | For efficient delivery of GFP fusion plasmids into mammalian cells for transient expression studies. |
| Anti-GFP Nanobody / Agarose Beads | Crucial for immunoprecipitation of the GFP fusion protein and its interaction partners from cell lysates. |
| Fluorescence-Compatible Cell Culture Medium | Phenol-red free medium for live-cell imaging to reduce background autofluorescence. |
| Confocal or High-Resolution Fluorescence Microscope | Essential for visualizing subcellular localization and potential co-localization of interacting proteins. |
Within the broader thesis investigating GFP fusion proteins for monitoring protein-protein interactions (PPIs), the choice of cellular host and the efficiency of delivering genetic constructs are foundational. This document provides detailed application notes and protocols for mammalian, yeast, and bacterial systems, focusing on their use in PPI studies using fluorescent protein tags. The optimal system balances physiological relevance, experimental tractability, and the specific requirements of the interaction being studied.
Application Notes
Mammalian Systems
Context for PPI Research: Essential for studying interactions in their native post-translational modification and compartmentalization context. GFP fusions (e.g., FRET, BiFC) are widely used for quantitative, real-time PPI analysis in living cells.
- Key Advantages: Native folding, correct compartmentalization, authentic PTMs.
- Key Limitations: Cost, slower growth, technical complexity.
- Common Cell Lines:
- HEK293T: High transfection efficiency, robust protein expression. Ideal for initial validation and protein production.
- HeLa: Well-characterized, suitable for cytoskeletal and signaling studies.
- CHO-K1: Preferred for stable line generation and scalable protein production.
Yeast Systems
Context for PPI Research: A powerful eukaryotic model offering genetics, speed, and simplicity. Yeast Two-Hybrid (Y2H) is a classic PPI discovery tool, while GFP fusions are used for localization and interaction studies via fluorescence microscopy or flow cytometry.
- Key Advantages: Genetic manipulability, rapid growth, cost-effectiveness for high-throughput screening.
- Key Limitations: Lack of mammalian PTMs, potential for non-native folding of mammalian proteins.
- Common Strains:
- S. cerevisiae: Standard for Y2H (e.g., AH109, Y2HGold strains) and fluorescence-based assays.
Bacterial Systems
Context for PPI Research: Primarily used for recombinant protein expression for in vitro PPI validation (e.g., co-purification, pull-down assays). GFP fusions can be used to monitor expression and solubility.
- Key Advantages: Highest yield, fastest growth, lowest cost.
- Key Limitations: No eukaryotic PTMs, inability to study membrane or compartmentalized proteins in their native state.
- Common Strains:
- E. coli BL21(DE3): Workhorse for T7-driven protein expression.
- E. coli DH5α: Standard for plasmid cloning and amplification.
Table 1: Comparison of Host Systems for GFP-PPI Research
| Feature | Mammalian (HEK293T) | Yeast (S. cerevisiae) | Bacterial (E. coli BL21) |
|---|---|---|---|
| Typical Transfection/Transformation Efficiency | 70-90% (Lipofection) | 10^3 - 10^5 CFU/µg DNA | 10^7 - 10^8 CFU/µg DNA |
| Doubling Time | ~24 hours | ~1.5 hours | ~20-30 minutes |
| Cost per Experiment | High | Low | Very Low |
| Protein Yield | Low to Moderate (mg/L) | Low (µg/L) | High (g/L) |
| Native Eukaryotic PTMs | Yes | Simple (Glycosylation, etc.) | No |
| Key PPI Assay Modality | FRET, BiFC, Co-IP | Yeast Two-Hybrid, Co-IP | Pull-down, Co-purification |
| Throughput Potential | Low to Moderate | High (Genetic screens) | High (Protein production) |
Experimental Protocols
Protocol 1: Transient Transfection of HEK293T Cells for FRET-based PPI Assay
Objective: To express GFP- and RFP-fusion protein constructs in mammalian cells for Förster Resonance Energy Transfer (FRET) measurement.
- Day 0: Seed HEK293T cells in a poly-D-lysine-coated 6-well plate at 3 x 10^5 cells/well in DMEM + 10% FBS. Incubate at 37°C, 5% CO2.
- Day 1: Prepare transfection complexes for each well:
- Dilute 2.5 µg total plasmid DNA (e.g., 1.25 µg GFP-X + 1.25 µg RFP-Y) in 250 µL Opti-MEM.
- Dilute 7.5 µL polyethylenimine (PEI, 1 mg/mL) in 250 µL Opti-MEM.
- Combine diluted PEI with diluted DNA, vortex, incubate 15-20 min at RT.
- Add dropwise to cells. Gently swirl plate.
- Day 2 (24h post-transfection): Replace media with fresh pre-warmed complete DMEM.
- Day 3 (48h post-transfection): Analyze cells. For FRET, harvest for microscopy or flow cytometry using appropriate filter sets for donor (GFP) emission and acceptor (RFP) sensitized emission.
Protocol 2: Yeast Transformation and Fluorescence Microscopy for GFP Fusion Localization
Objective: To introduce a GFP-fusion plasmid into yeast for protein localization analysis.
- Inoculate a single colony of desired yeast strain (e.g., BY4741) in 5 mL YPD. Grow overnight at 30°C, 250 rpm.
- Day 2: Dilute overnight culture to OD600 ~0.2 in fresh YPD. Grow to OD600 ~0.8 (mid-log phase).
- Harvest 1.5 mL cells by centrifugation (3000 x g, 5 min). Wash with 1 mL sterile water.
- Resuspend pellet in 240 µL transformation mix: 240 µL 50% PEG-3350, 36 µL 1M LiAc, 50 µL 2 mg/mL carrier DNA (denatured), 34 µL sterile H2O, and 5 µL plasmid DNA (0.1-1 µg).
- Vortex vigorously, incubate at 42°C for 40 minutes.
- Pellet cells (3000 x g, 30 sec), resuspend in 500 µL sterile H2O. Plate 100-200 µL on appropriate selective medium (e.g., -Leu). Incubate at 30°C for 2-3 days.
- Day 4/5: Pick a colony, grow in selective medium to log phase. Mount 5 µL culture on a slide. Image GFP fluorescence using a microscope with a standard FITC/GFP filter set.
Protocol 3: Recombinant Protein Expression of a GFP-Fusion in E. coli
Objective: To express and purify a GFP-fusion protein for in vitro interaction studies.
- Transform expression plasmid (e.g., pET-28a-GFP-X) into chemically competent E. coli BL21(DE3). Plate on LB agar with appropriate antibiotic (e.g., Kanamycin). Incubate overnight at 37°C.
- Day 2: Inoculate a single colony into 5 mL LB+Kan. Grow overnight at 37°C, 250 rpm.
- Day 3: Dilute 1:100 into 100 mL fresh LB+Kan in a baffled flask. Grow at 37°C, 250 rpm to OD600 ~0.6.
- Induce protein expression by adding IPTG to a final concentration of 0.5 mM.
- Shift temperature to 18°C and incubate with shaking for 16-20 hours (overnight).
- Day 4: Harvest cells by centrifugation (4,000 x g, 20 min, 4°C). Proceed with cell lysis (e.g., sonication in lysis buffer) and purification via affinity chromatography (e.g., Ni-NTA resin for His-tagged GFP-fusion). Monitor purity by SDS-PAGE and GFP fluorescence.
Visualizations
Title: Host System Selection Workflow for GFP-PPI Studies
Title: FRET Principle for Detecting Protein-Protein Interactions
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for GFP-PPI Experiments
| Reagent / Material | Function in GFP-PPI Research | Example Product/Brand |
|---|---|---|
| Polyethylenimine (PEI) | Cationic polymer for transient transfection of mammalian cells; cost-effective for large-scale DNA delivery. | Linear PEI (Polysciences), PEI MAX. |
| Lipofectamine 3000 | Lipid-based transfection reagent for high-efficiency, low-toxicity delivery of DNA/RNA into mammalian cells. | Thermo Fisher Scientific. |
| FuGENE HD | Non-liposomal transfection reagent; low cytotoxicity, effective in serum-containing media. | Promega. |
| LiAc/PEG Mix | Chemical transformation mixture for yeast; enables plasmid DNA uptake through cell wall permeabilization. | Homemade (LiAc, PEG-3350, SS-DNA) or commercial kits. |
| Chemically Competent E. coli | Bacteria treated for efficient plasmid uptake via heat shock, essential for cloning and protein expression. | DH5α (cloning), BL21(DE3) (expression). |
| Protease Inhibitor Cocktail | Prevents degradation of GFP-fusion proteins and their interaction partners during cell lysis and purification. | cOmplete (Roche), PMSF, Pepstatin A. |
| Ni-NTA Agarose | Affinity resin for purification of polyhistidine (His)-tagged GFP-fusion proteins from bacterial/yeast lysates. | Qiagen, Thermo Fisher Scientific. |
| Anti-GFP Nanobody/Antibody | For immunoprecipitation (Co-IP) of GFP-fusion proteins and their interacting partners. | GFP-Trap (Chromotek), Anti-GFP mAb. |
| FRET Filter Sets | Specialized microscope filter cubes to isolate donor emission and acceptor sensitized emission for FRET imaging. | CFP/YFP FRET set, GFP/RFP FRET set. |
Within the broader thesis investigating protein-protein interactions (PPIs) using GFP-based fusion proteins, the selection and optimization of imaging hardware and software is paramount. Techniques like Fluorescence Resonance Energy Transfer (FRET), Bimolecular Fluorescence Complementation (BiFC), and rationetric analysis provide critical spatial and temporal data on PPIs but impose distinct and stringent demands on the microscopy system. This application note details the specific microscopy requirements and experimental protocols for these three cornerstone techniques, ensuring data robustness and reproducibility in dynamic PPI research.
Core Microscopy System Requirements
Successful implementation of FRET, BiFC, and rationetric imaging hinges on a well-configured system. The key components are summarized below.
Table 1: Core Microscope Components & Specifications
| Component | FRET (e.g., FRET-FLIM or Acceptor Photobleaching) | BiFC | Rationetric Analysis (e.g., pH, Ca²⁺ indicators) | Rationale |
|---|---|---|---|---|
| Light Source | High-intensity laser (FLIM) or stable Xenon/Metal Halide lamp. | Standard LED or metal halide lamp. | Stable, flicker-free light source (LED preferred). | FRET-FLIM requires pulsed lasers; intensity stability is critical for quantitative ratio imaging. |
| Objective Lens | High NA (≥1.4) oil-immersion, Plan-Apochromat. | Plan-Apochromat (40x-63x), high NA. | Plan-Apochromat, high transmission. | Maximizes signal collection and spatial resolution. |
| Filter Sets / Monochromators | Critical: Precise donor/acceptor emission separation. Fast filter wheels or tunable monochromators. | Standard GFP/YFP filter sets. | Critical: Matched filter sets for two emission wavelengths with minimal crosstalk. | Accurate spectral separation is non-negotiable for FRET and rationetric quantification. |
| Detector | FLIM: High-speed photon-counting PMT. Intensity: sCMOS or EMCCD with high quantum yield. | sCMOS or EMCCD. | sCMOS with high linearity and dynamic range. | Sensitivity, speed, and quantitative linearity are essential. |
| Environmental Control | Live-cell chamber (37°C, 5% CO₂). | Live-cell chamber. | Live-cell chamber, precise temperature control. | Maintains physiological conditions for all live-cell PPI studies. |
| Software | Capable of spectral unmixing, kinetic analysis, FLIM fitting, and ratio image calculation. | Time-lapse acquisition, background subtraction. | Real-time ratio calculation (F₁/F₂), calibration curve integration. | Enables accurate data processing and visualization. |
Table 2: Key Filter Specifications for Common Fluorophore Pairs
| Technique | FP Pair | Excitation (nm) | Dichroic (nm) | Emission 1 (nm) | Emission 2 (nm) |
|---|---|---|---|---|---|
| FRET | CFP/YFP (e.g., Cerulean/Venus) | 430-450 (CFP) | 455-510 | 460-500 (Donor) | 520-550 (Acceptor) |
| FRET | GFP/mCherry | 460-490 (GFP) | 495-550 | 500-550 (Donor) | 570-620 (Acceptor) |
| BiFC | Venus (reconstituted) | 500-520 | 515-550 | 520-550 | N/A |
| Rationetric | pHluorin (rationetric) | 400 & 480 (Dual) | 450-490 | 500-550 | 500-550 (Intensity Ratio) |
Detailed Experimental Protocols
Protocol 1: FRET by Acceptor Photobleaching (Microscopy Setup Validation)
Principle: Selective photodestruction of the acceptor fluorophore (e.g., mCherry) should increase the donor (e.g., GFP) fluorescence if FRET occurs.
- Sample Prep: Seed cells expressing GFP- and mCherry-tagged proteins of interest on imaging dishes.
- Image Acquisition: Using a confocal or widefield system:
- Define a Region of Interest (ROI).
- Acquire donor (GFP) and acceptor (mCherry) pre-bleach images using minimal laser power to avoid bleaching.
- Bleach the acceptor in the ROI using high-power 561nm laser illumination (100% power, 5-10 iterations).
- Re-acquire post-bleach donor and acceptor images using the pre-bleach settings.
- Analysis: Calculate FRET efficiency: E = 1 - (FDpre / FDpost), where F_D is donor intensity. A positive efficiency indicates FRET.
Protocol 2: Bimolecular Fluorescence Complementation (BiFC) Assay
Principle: Two non-fluorescent fragments of a fluorescent protein (e.g., Venus) are fused to putative interacting proteins. Interaction drives complementation and fluorescence.
- Transfection: Co-transfect cells with plasmids encoding proteins X-VN173 and Y-VC155 (Venus N- and C-terminal fragments).
- Controls: Include pairs with known interaction (positive) and non-interacting proteins (negative).
- Imaging: 48-72 hours post-transfection, image using standard YFP filters.
- Use identical exposure times and lamp power between samples.
- Focus on cytoplasmic signal; nuclear fluorescence can be nonspecific.
- Analysis: Quantify fluorescence intensity in the reconstituted channel. Signal significantly above the negative control indicates interaction. Note: BiFC is irreversible.
Protocol 3: Live-Cell Rationetric Calcium Imaging using GCaMP-RG
Principle: The GCaMP-RG indicator exhibits a calcium-dependent emission shift, allowing ratio-metric quantification.
- Dye/Loading: Transfect cells with plasmid encoding GCaMP-RG.
- Setup: Configure microscope for dual-emission ratio imaging:
- Excitation: 488 nm (common for both forms).
- Emission: Collect light simultaneously at 510nm (F510) and 405nm (F405) using a beamsplitter or rapid filter wheel.
- Acquisition: Establish a stable baseline (≥30 sec), then apply stimulus. Acquire images at 1-5 sec intervals.
- Processing: For each time point, create a ratio image (R = F510 / F405). This ratio is independent of indicator concentration and photobleaching. Convert ratio to [Ca²⁺] using an in situ calibration curve.
Diagram: Experimental Workflow for PPI Techniques
Title: Workflow for PPI Imaging Techniques
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for GFP-Fusion PPI Imaging
| Reagent/Material | Function & Role in Experiment | Example/Vendor |
|---|---|---|
| Validated FP-Fusion Plasmids | Ensure proper folding and function of the fused protein; backbone determines brightness, oligomerization. | Addgene repository vectors: pEGFP-N1, pmCherry-C1, Venus BiFC fragments (VC155/VN173). |
| High-Fidelity Transfection Reagent | Deliver plasmid DNA into cells with high efficiency and low cytotoxicity for robust expression. | Lipofectamine 3000 (Thermo), FuGENE HD (Promega), or polyethylenimine (PEI). |
| Live-Cell Imaging Media | Maintain pH, osmolarity, and nutrients without fluorescence background during time-lapse. | Phenol-red free DMEM with HEPES or CO₂-independent medium. |
| Immobilization Reagents | Anchor cells for stable, long-term imaging without affecting membrane integrity. | Poly-D-Lysine, Cell-Tak (Corning), or µ-Slide dishes (ibidi). |
| Spectral Control Plasmids | Express donor or acceptor alone for correcting spectral bleed-through (SBT) in FRET. | CFP-only and YFP-only plasmids for calibration. |
| Commercial FRET/ BiFC Kits | Provide optimized, validated positive and negative control pairs for assay standardization. | Lonza BiFC Kit, FP-based FRET biosensors (e.g., CKAR). |
| Environmental Chamber | Maintain precise temperature (37°C) and CO₂ (5%) for physiological live-cell imaging. | Stage-top incubators (Tokai Hit, OkoLab). |
| Calibration Standards | Create in situ calibration curves for rationetric indicators (e.g., high/low pH, Ca²⁺ buffers). | Ionophore cocktails (e.g., Ionomycin, Nigericin) with defined buffers. |
Application Notes: GFP-Based Assays in Signaling & Drug Discovery
The integration of GFP-fusion proteins into signaling pathway research provides a dynamic window into cellular processes, enabling both basic mechanistic discovery and applied pharmaceutical development. These tools are pivotal for target identification, validation, and the characterization of drug mechanisms of action (MoA).
1.1. Target Identification & Validation Using GFP Reporters Genetically encoded GFP reporters under the control of pathway-specific response elements (e.g., NF-κB, AP-1, SRE, HIF) allow for high-throughput screening of compound libraries. Activation or inhibition of the pathway is quantified via fluorescence intensity. Furthermore, endogenous gene tagging with GFP via CRISPR/Cas9 facilitates the study of native protein localization and abundance without overexpression artifacts, confirming target engagement in physiologically relevant models.
1.2. Monitoring Protein-Protein Interactions (PPIs) For direct PPI monitoring within signaling cascades, techniques like Fluorescence Resonance Energy Transfer (FRET) and Bimolecular Fluorescence Complementation (BiFC) using spectral variants of GFP (e.g., CFP/YFP) are standard. These methods are critical for validating interactions between drug targets (e.g., kinases) and their substrates or regulatory proteins. Disruption of FRET signal by a small molecule provides direct evidence of PPI inhibition.
1.3. Quantitative Data from Key Assays The following table summarizes core quantitative parameters for GFP-based assays in drug discovery contexts.
Table 1: Quantitative Parameters for GFP-Based Signaling & Drug Discovery Assays
| Assay Type | Primary Readout | Typical Z'-Factor | Throughput | Key Application in Drug Discovery |
|---|---|---|---|---|
| GFP Reporter Gene | Fluorescence Intensity | 0.5 - 0.7 | High (HTS) | Primary screening for pathway modulators |
| FRET (CFP/YFP) | Donor/Acceptor Emission Ratio | 0.3 - 0.6 | Medium | Target engagement & PPI inhibition |
| FLIP/FRAP (GFP-Fusion) | Recovery Half-life (t₁/₂) | N/A (Kinetic) | Low | Measuring protein turnover & complex stability |
| CRISPR-GFP Endogenous Tag | Localization/Intensity | N/A (Image-based) | Medium | Target validation & phenotypic screening |
1.4. Case Study: EGFR Signaling Pathway The Epidermal Growth Factor Receptor (EGFR) pathway is a prime example. GFP-tagged Grb2 or SOS proteins can visualize recruitment to activated EGFR at the membrane via TIRF microscopy. FRET between GFP-EGFR and YFP-SH2 domains confirms receptor autophosphorylation. Drug candidates (e.g., tyrosine kinase inhibitors, TKIs) are rapidly validated by their dose-dependent inhibition of these GFP-visible events, linking biochemical inhibition to cellular phenotype.
Experimental Protocols
Protocol 2.1: FRET Assay for Monitoring Kinase-Substrate Interaction Disruption Objective: To test a compound's ability to disrupt the interaction between a GFP-tagged kinase and a YFP-tagged substrate in live cells.
Materials:
- HEK293T or relevant cell line
- Plasmids: pEGFP-N1-Kinase, pEYFP-N1-Substrate
- Transfection reagent (e.g., PEI)
- Putative inhibitory compound(s)
- DMSO (vehicle control)
- 96-well glass-bottom plate
- Microplate reader or confocal microscope with FRET filters
Procedure:
- Seed & Transfect: Seed cells in a 96-well plate at 50% confluence. Co-transfect with GFP-Kinase and YFP-Substrate plasmids (1:1 ratio, 200 ng total DNA/well) using standard protocols.
- Compound Treatment: 24h post-transfection, treat cells with a dose range of the test compound (e.g., 0.1 nM - 10 µM) or DMSO control for 4-6 hours.
- FRET Imaging/Acquisition:
- On a plate reader: Acquire fluorescence using CFP excitation (433 nm) and measure emissions at 475 nm (CFP channel) and 527 nm (FRET channel).
- On a microscope: Use a 458 nm laser line and collect emissions with 470/30 nm (CFP) and 535/30 nm (FRET) bandpass filters.
- Data Analysis:
- Calculate FRET Ratio (FRET channel emission / CFP channel emission) for each well.
- Normalize FRET ratios to the DMSO control (set as 100% interaction).
- Plot dose-response curve. A significant decrease in FRET ratio indicates disruption of the kinase-substrate interaction.
Protocol 2.2: CRISPR/Cas9-Mediated Endogenous GFP Tagging for Target Validation Objective: To tag the native locus of a signaling protein (e.g., IκBα) with GFP to monitor its dynamics in response to pathway stimulation and inhibition.
Materials:
- Cell line of interest
- pSpCas9(BB)-2A-GFP (PX458) plasmid
- Donor plasmid: Contains homology arms (800 bp) flanking the STOP codon of the target gene, followed by a linker and GFP sequence.
- sgRNA targeting sequence near the STOP codon
- Puromycin or appropriate selection antibiotic
- Flow sorter
Procedure:
- Design & Cloning: Design sgRNA targeting the genomic region immediately upstream of the target gene's STOP codon. Clone this sequence into PX458. Construct the homologous donor plasmid.
- Co-transfection: Co-transfect cells with the PX458-sgRNA plasmid and the donor plasmid.
- Selection & Sorting: 48-72h post-transfection, apply puromycin selection for 3-5 days. Harvest cells and use FACS to sort the top 5-10% GFP-positive population into a 96-well plate for clonal expansion.
- Validation: Screen clones by PCR and Western blot for correct GFP tagging (size shift). Validate functionally by stimulating the relevant pathway (e.g., with TNFα for NF-κB) and monitoring GFP-IκBα degradation via live-cell imaging or immunoblot.
- Application: Use validated clone for compound screening to identify stabilizers of IκBα that would inhibit NF-κB activation.
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for GFP-Based PPI & Signaling Studies
| Reagent / Material | Function & Application |
|---|---|
| pEGFP/N1/C1 Vector Series | Standard plasmids for generating N- or C-terminal GFP fusion proteins via cloning. |
| sGFP2 & mNeonGreen | Next-generation, brighter, and more photostable GFP variants for superior imaging. |
| CRISPR/Cas9 GFP Donor Vectors (e.g., pDDonor-GFP) | Ready-to-use templates with homology arms for endogenous gene tagging via HDR. |
| FRET Standards (mCerulean3/mVenus) | Optimized, inert CFP/YFP FRET pair with high quantum yield for reliable calibration. |
| HaloTag-GFP Ligands | Enables pulse-chase labeling and multi-color imaging of different protein pools. |
| NanoBIT PPI System | Complementation-based system using small fragments of luciferase; highly sensitive for weak/transient interactions. |
| Photoactivatable GFP (paGFP) | Allows selective activation in a region of interest to study protein diffusion and trafficking. |
| CellLight Reagents (BacMam) | Pre-made, ready-to-use baculovirus for expressing GFP-tagged cellular structures (e.g., GFP-Actin). |
Visualizations
Title: GFP Tools Map Signaling from Receptor to Drug Readout
Title: GFP-PPI Drug Screening Experimental Workflow
Solving Common Problems: Optimization and Troubleshooting for GFP-PPI Experiments
Within the broader thesis on utilizing GFP fusions for monitoring protein-protein interactions (PPIs), managing background fluorescence is a critical determinant of experimental success. High background obscures specific signals, reduces the signal-to-noise ratio (SNR), and compromises the quantification of dynamic interactions. This document details the principal causes and provides validated, actionable protocols for background reduction.
Causes of High Background Fluorescence
Background fluorescence in GFP-based PPI studies arises from multiple sources, categorized below.
Table 1: Primary Causes of High Background Fluorescence
| Category | Specific Cause | Typical Impact on SNR Reduction |
|---|---|---|
| Sample Autofluorescence | NAD(P)H, Flavins, Collagen, Lipofuscin | 20-50% |
| Non-Specific Probe Binding | Hydrophobic interactions, Ionic interactions | 30-70% |
| Cellular Stress & Fixation Artifacts | Aldehyde-induced fluorescence, pH shifts, ROS generation | 40-80% |
| Optical & Instrument Factors | Camera read noise, Stray light, Imperfect filter sets | 15-40% |
| Expression & Biological Factors | Free GFP/untagged protein, Protein aggregation, Overexpression | 50-90% |
Research Reagent Solutions Toolkit
Table 2: Essential Reagents for Background Reduction in GFP-PPI Studies
| Reagent / Material | Function | Example Product/Catalog |
|---|---|---|
| Tissue Culture Media (Phenol Red-free) | Eliminates media-derived autofluorescence. | Gibco FluoroBrite DMEM |
| Quenching / Reducing Agents | Reduces aldehyde-induced fluorescence post-fixation. | 0.1% Sodium Borohydride (NaBH4) |
| Blocking Buffers | Minimizes non-specific antibody/probe binding. | 5% BSA in TBST, 10% Normal Goat Serum |
| Commercial Mounting Media with Antifade | Preserves fluorescence, reduces photobleaching. | ProLong Diamond, VECTASHIELD |
| Protease Inhibitor Cocktail | Prevents GFP tag cleavage & free GFP accumulation. | cOmplete, EDTA-free (Roche) |
| Live-Cell ROS Scavengers | Lowers oxidative stress-induced autofluorescence. | 1-5 mM Ascorbic Acid |
| High-Affinity, Validated Antibodies | Ensures specific immunodetection in IF protocols. | Chromotek GFP-Trap Agarose |
| Plasmid with Degron-Tagged GFP | Enables rapid degradation of unbound fusion protein. | dTAG or HaloTag systems |
Protocols for Background Reduction
Protocol 1: Minimizing Autofluorescence in Live-Cell Imaging
Objective: Prepare live cells for GFP-PPI imaging with minimal intrinsic background.
- Culture Cells: Grow cells expressing the GFP-fusion protein of interest in phenol red-free medium for ≥24h before imaging.
- Media Exchange: 30 min prior to imaging, replace medium with pre-warmed, CO₂-equilibrated imaging medium (e.g., FluoroBrite DMEM + 2% FBS + 25mM HEPES).
- ROS Scavenger Treatment (Optional): For sensitive cells, add 1 mM sodium ascorbate to the imaging medium.
- Incubation: Maintain cells at 37°C/5% CO₂ until immediate transfer to the microscope stage-top incubator.
- Image Acquisition: Use the minimum laser power and exposure time that yield a detectable specific signal. Acquire a control image from an untransfected cell under identical settings to quantify background.
Protocol 2: Post-Fixation Quenching for Immunofluorescence
Objective: Eliminate autofluorescence induced by paraformaldehyde (PFA) fixation.
- Fix Cells: Fix cells expressing the GFP-fusion protein with 4% PFA for 15 min at RT.
- Wash: Rinse 3x with 1X PBS, 5 min per wash.
- Prepare Quenching Solution: Freshly prepare a 0.1% (w/v) Sodium Borohydride (NaBH4) solution in 1X PBS. Caution: Hydrogen gas evolution.
- Quench: Incubate cells with the NaBH4 solution for 10 min at RT.
- Wash: Rinse thoroughly with 1X PBS, 4 times for 10 min each, to remove all residues.
- Proceed: Continue with standard immunofluorescence protocol (blocking, antibody staining).
Protocol 3: Optimized Immunoprecipitation for Free GFP Removal
Objective: Isolate specific protein complexes while removing free GFP or aggregated fusion protein.
- Lysis: Lyse cells expressing the GFP-fusion protein in ice-cold lysis buffer (e.g., 50mM Tris-HCl pH 7.5, 150mM NaCl, 1% NP-40, 1x protease inhibitors) for 30 min at 4°C.
- Clarification: Centrifuge at 16,000 x g for 15 min at 4°C. Transfer supernatant to a new tube.
- Pre-Clear: Incubate lysate with 20 µL of bare agarose resin for 30 min at 4°C. Centrifuge to collect supernatant.
- GFP-Trap Incubation: Incubate pre-cleared lysate with 20 µL of equilibrated GFP-Trap Agarose beads for 2h at 4°C with gentle rotation.
- Wash: Pellet beads (2,500 x g, 2 min) and wash 4x with 500 µL of ice-cold lysis buffer.
- Elute: Elute bound complexes with 40 µL of 2X Laemmli SDS sample buffer by heating at 95°C for 5 min. Analyze by Western Blot.
Visualizations
Title: Sources of High Fluorescence Background
Title: Workflow for Background Reduction Strategies
Within the context of a thesis on GFP fusion proteins for monitoring protein-protein interactions (PPIs), distinguishing genuine interactions from experimental artifacts is paramount. The overexpression inherent in many GFP-based assays (e.g., FRET, Co-IP, BiFC) can lead to two critical issues: (1) False Positives from non-specific interactions and mislocalization due to unnaturally high protein concentrations, and (2) False Negatives from epitope masking, improper folding, or disrupted native complex formation. These Application Notes provide detailed protocols and controls to validate PPI data, emphasizing quantitative rigor and reproducibility for researchers and drug development professionals.
The following table summarizes common artifacts, their mechanistic causes, and recommended validation controls with associated quantitative benchmarks.
Table 1: Artifacts, Causes, and Validation Controls in GFP-PPI Assays
| Artifact Type | Primary Cause | Potential Consequence | Recommended Control Experiment | Target Metric / Benchmark |
|---|---|---|---|---|
| Non-Specific Interaction (False Positive) | High local concentration from overexpression; hydrophobic or charged surface patches. | Spurious, non-physiological binding. | Expression Titration | Linear correlation (R² >0.9) of signal with expression level of lower-abundance partner. |
| Crowding/Mislocalization (False Positive) | Saturation of binding sites or native organelles; altered subcellular trafficking. | Ectopic colocalization not reflective of native state. | Endogenous Tagging Comparison | >70% colocalization coefficient between GFP-fusion and endogenously tagged protein (e.g., via CRISPR). |
| Competitive Disruption (False Negative) | GFP tag or linker interferes with binding interface; steric hindrance. | Loss of known interaction. | Tag Swapping & Truncation | Interaction recovery >80% with tag on opposite terminus or minimal tag (e.g., 11-aa tag). |
| Fluorescence Resonance Energy Transfer (FRET) Bleed-Through (False Positive) | Direct excitation of acceptor or donor emission spectral overlap. | Apparent FRET without proximity. | Acceptor & Donor-Only Controls | FRET efficiency <2% in control cells. Corrected NFRET used for analysis. |
| Biomolecular Fluorescence Complementation (BiFC) Self-Assembly (False Positive) | Spontaneous, irreversible reassembly of split fluorescent protein fragments. | Signal without target protein interaction. | Fragment Pairing Controls (e.g., fused to non-interacting proteins, cytosolic localization) | Signal intensity <10% of experimental pair. |
| Altered Proteostasis (False Positive/Negative) | Overexpression overwhelms folding or degradation machinery; aggregates. | Toxic phenotypes; aggregation-induced co-recruitment. | Solubility & Localization Checks (with counterstains) | >90% of fluorescence in soluble fraction; coherent localization pattern. |
Detailed Experimental Protocols
Protocol 1: Expression Titration for Specificity Validation
Objective: To establish that the observed PPI signal is dependent on the specific affinity between partners and not merely on the concentration of the overexpressed proteins.
Materials:
- Plasmid for GFP-tagged Protein A (GFP-A).
- Plasmid for RFP/mCherry-tagged Protein B (RFP-B).
- A null/empty vector plasmid or a plasmid expressing a scrambled protein.
- Transfection reagent suitable for your cell line.
- Controlled cell line (e.g., HEK293T, HeLa).
- Flow cytometer or fluorescence microscope with quantitative imaging software.
Procedure:
- Experimental Design: Seed cells in a 12-well plate. Prepare a constant amount of GFP-A plasmid (e.g., 0.5 µg) for each transfection.
- Titrate Protein B: Co-transfect the constant GFP-A with a gradient of RFP-B plasmid (e.g., 0.05, 0.1, 0.25, 0.5, 1.0 µg). For the crucial control, include a point where RFP-B is replaced by an equal mass of empty vector or non-interacting RFP-control plasmid.
- Harvest & Analyze: 24-48h post-transfection, harvest cells. Analyze using:
- Flow Cytometry: Gate on double-positive (GFP+/RFP+) cells. Plot the median FRET ratio (or co-localization coefficient from imaging) against the median RFP intensity (proxy for Protein B expression level).
- Interpretation: A specific, saturable interaction will show a hyperbolic curve approaching a plateau. A linear, non-saturating relationship suggests the signal is heavily driven by overexpression. The control transfections should yield a near-zero, flat line.
Protocol 2: Acceptor Photobleaching FRET (apFRET) with Essential Controls
Objective: To measure genuine FRET while correcting for spectral bleed-through (SBT), a major source of false positives.
Materials:
- Cells expressing GFP (donor) and RFP (acceptor) fusion proteins.
- Confocal or epifluorescence microscope with a photobleaching module and precise region-of-interest (ROI) selection.
- Appropriate filter sets for GFP and RFP.
Procedure:
- Sample Preparation: Include three mandatory control samples alongside experimental (GFP-A + RFP-B):
- Donor Only: Cells expressing GFP-A alone.
- Acceptor Only: Cells expressing RFP-B alone.
- Free FP Control: Cells co-expressing unfused GFP and unfused RFP.
- Image Acquisition:
- Define a bleach ROI on a cell expressing both fluorophores.
- Acquire a pre-bleach donor (GFP) image and acceptor (RFP) image.
- Bleach the acceptor in the ROI using high-intensity RFP excitation light.
- Acquire a post-bleach donor image and acceptor image (to confirm bleaching).
- SBT Correction & Calculation:
- Measure donor intensity in the bleached ROI pre- (IDpre) and post-bleach (IDpost).
- Calculate uncorrected FRET efficiency: E = 1 - (IDpre / IDpost).
- Using the Donor Only sample, calculate the SBT of donor emission into the acceptor channel. Using the Acceptor Only sample, calculate the direct excitation of the acceptor by the donor laser line.
- Apply these SBT correction factors to the experimental images to generate corrected donor intensities before calculating final E.
- Interpretation: Genuine FRET is indicated by a significant increase in donor fluorescence after acceptor bleaching (>5-10% after SBT correction). The free FP control should yield E ≈ 0%.
Visualizing Workflows and Relationships
Title: Decision Workflow for Validating GFP-PPI Results
Title: Overexpression Artifacts Leading to False PPI Signals
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Controlling GFP-PPI Experiments
| Reagent / Material | Function & Purpose in Controlling Artifacts | Example Product/Catalog |
|---|---|---|
| Tunable Expression Vectors | Enables precise titration of protein expression levels (e.g., via inducible promoters or varying plasmid dose) to test for concentration-dependent artifacts. | Tet-On 3G Inducible System; low-copy number vectors (e.g., pRS series). |
| Fluorescent Protein Variant Pairs (Optimized for FRET) | Pre-validated donor/acceptor pairs with minimal spectral bleed-through (e.g., mNeonGreen/iFluor604, mCerulean3/mVenus). Reduce SBT false positives. | mNeonGreen-iFluor604 FRET pair; mTurquoise2-YPet. |
| Split-FP Negative Control Plasmids | Vectors expressing non-interacting protein fusions for BiFC or split-GFP assays. Essential baseline for spontaneous fragment assembly. | pBiFC-bJun-cFos (positive) & pBiFC-bJun-cFos(mut) (negative) vectors. |
| CRISPR/Cas9 Knock-in Reagents | For creating endogenously tagged cell lines, avoiding overexpression entirely. Gold standard for localization and interaction validation. | Synthetic crRNA/tracrRNA; donor homology templates for N-/C-terminal tagging. |
| Proteostasis Interference Controls | Small molecule inhibitors (e.g., proteasome, autophagy) or co-transfected chaperones to test if PPI signal depends on healthy protein folding/degradation. | MG132 (proteasome inhibitor); Bafilomycin A1 (autophagy inhibitor); Hsp70 co-expression plasmid. |
| Microscopy Calibration Beads | Multi-spectral beads for aligning channels and correcting for optical crosstalk in colocalization and FRET imaging workflows. | TetraSpeck Microspheres. |
| Mild, Reversible Crosslinkers | Used prior to Co-IP of weak/transient interactions, but can also induce false positives; must be titrated carefully against no-crosslink control. | Dithiobis(succinimidyl propionate) (DSP); Formaldehyde. |
Thesis Context: This application note, within a broader thesis on GFP fusions for monitoring protein-protein interactions (PPIs), details strategies to overcome low signal-to-noise ratio (SNR) in fluorescence microscopy. Optimizing fluorophore pairs and optical filter sets is critical for detecting weak interaction signals, such as those from Förster Resonance Energy Transfer (FRET), against cellular autofluorescence and background noise.
Monitoring PPIs via GFP-family fusions often involves subtle spectral changes (e.g., FRET, spectral shift biosensors) where the signal of interest is a small fraction of the total emitted light. A low SNR obscures these changes, leading to false negatives or poor quantification. Key noise sources include:
- Cellular Autofluorescence: From NAD(P)H, flavins, and lipofuscin.
- Direct Excitation: Of the acceptor fluorophore by the donor excitation light.
- Bleed-Through (Crosstalk): Donor emission detected in the acceptor channel, and vice versa.
- Photodetector Noise: Especially critical in low-light conditions.
- Non-Specific Background Fluorescence.
Quantitative Comparison of Modern Fluorophore Pairs
Optimal fluorophore pairing maximizes Förster distance (R₀), minimizes crosstalk, and matches available filter sets.
Table 1: Characteristics of Optimized Fluorescent Protein Pairs for FRET
| Donor | Acceptor | R₀ (nm) | Donor Ex/Em (nm) | Acceptor Ex/Em (nm) | Primary Advantage | Best Suited For |
|---|---|---|---|---|---|---|
| mCerulean3 | mVenus | 5.4 | 433/475 | 515/528 | High quantum yield, mature fast | General cytosolic PPIs |
| mTurquoise2 | mNeonGreen | 6.2 | 434/474 | 506/517 | Very bright, high R₀, photostable | Low-expression interactions |
| mScarlet-I | mNeonGreen | 4.9 | 569/594 | 506/517 | Red-shifted, reduces autofluorescence | Tissues or high-autofluorescence cells |
| Clover | mRuby2 | 5.7 | 486/506 | 559/600 | Large Stokes shift, reduces crosstalk | Ratiometric biosensors |
| mTurquoise2 | sREACh | 6.8* | 434/474 | Ex: Donor FRET | Exceptional R₀, zero direct acceptor excitation | Quantifying very weak or transient PPIs |
*R₀ for mTurquoise2/sREACh is an engineered variant optimized for acceptor excitation via FRET only.
Optical Filter Set Optimization Protocol
The choice of filter sets (excitation, emission, dichroic) is as crucial as fluorophore selection.
Protocol 3.1: Filter Set Selection and Validation for FRET Objective: To configure and validate microscope filter sets for maximum SNR in a FRET experiment using mTurquoise2 (donor) and mNeonGreen (acceptor).
Materials:
- Microscope with configurable filter cubes or tunable filter system.
- Cells expressing donor-only, acceptor-only, and donor-acceptor fusion constructs.
- High-quality bandpass filters and multi-band dichroic mirrors.
Procedure:
- Donor Excitation/Acceptor Emission (Sensitized Emission) Channel Setup:
- Use a donor excitation filter (e.g., 430/24 nm BP).
- Select a dichroic mirror that reflects ≤450 nm and transmits ≥470 nm.
- For the emission filter, choose a narrow bandpass centered on the acceptor's peak emission (e.g., 525/40 nm BP for mNeonGreen). This minimizes donor bleed-through.
- Crosstalk Calibration Imaging:
- Image the donor-only sample using the FRET filter set. The signal here is donor bleed-through (BTD). Record intensity.
- Image the acceptor-only sample using the FRET filter set. The signal here is direct excitation of the acceptor (DEA). Record intensity.
- SNR Calculation & Optimization:
- Image the FRET sample. The raw FRET signal (Fraw) contains true FRET (F), BTD, and DEA.
- Calculate corrected FRET:
F = F<sub>raw</sub> - (BT<sub>D</sub> + DE<sub>A</sub>). - Measure background (B) from a cell-free region. Signal-to-Noise Ratio is approximated as:
SNR = F / √(F + B). - Iterate: If SNR is low (<3), try narrower emission bandpass filters or consider a more spectrally separated fluorophore pair.
Diagram 1: FRET Filter Configuration for SNR
Detailed Protocol: FRET Efficiency Measurement with Optimized SNR
Protocol 4.1: Acceptor Photobleaching FRET Assay Objective: To measure FRET efficiency (E) by selectively destroying the acceptor fluorophore, providing a robust SNR-independent metric.
Materials:
- Confocal or widefield microscope with a focused, high-intensity laser or light source for bleaching at the acceptor's excitation peak.
- Cells expressing the donor-acceptor fusion protein.
- Software for region-of-interest (ROI) analysis and image arithmetic.
Procedure:
- Pre-bleach Acquisition:
- Focus on a cell expressing the fusion construct.
- Using the donor channel (excite donor, collect donor emission), acquire an image (ID_pre).
- Using the acceptor channel, acquire an image to confirm acceptor presence.
- Acceptor Photobleaching:
- Define a precise ROI within the cell.
- Switch to high-power acceptor excitation light (e.g., 514 nm laser at 100% power).
- Bleach the ROI until acceptor fluorescence is reduced by >80% (monitor via acceptor channel).
- Post-bleach Acquisition:
- Immediately re-acquire the donor channel image (ID_post) using identical settings as step 1.
- Calculation & Analysis:
- Calculate FRET Efficiency E:
E = 1 - (I<sub>D_pre</sub> / I<sub>D_post</sub>). - A positive E (increase in donor fluorescence after acceptor loss) indicates true FRET. The magnitude of E reflects the proximity/interaction strength.
- SNR Note: This method inherently improves SNR for the measurement of E because the signal change (ΔID) is specific to the bleached ROI, and background noise is subtracted during ratio calculation.
- Calculate FRET Efficiency E:
Diagram 2: Acceptor Photobleaching FRET Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for High-SNR PPI Imaging
| Item | Function & Relevance to SNR | Example/Note |
|---|---|---|
| Genetically Encoded Biosensors | Provide a direct, ratiometric readout of PPI or activity, reducing variability and improving functional SNR. | MP-p38 biosensor: Uses differentially colored FPs to map kinase activity. |
| Tandem Purification Tags | Allows precise control of expression levels via affinity purification, reducing non-specific aggregation noise. | Strep-II/6xHis: For purifying FP-fusion proteins to validate stoichiometry. |
| Live-Cell Imaging Media | Phenol-red free, low-fluorescence media to minimize background and autofluorescence. | FluoroBrite DMEM |
| Mounting Reagents with Anti-fade | For fixed samples, reduces photobleaching, allowing longer exposures for better SNR without signal loss. | Prolong Diamond Antifade Mountant |
| Plasmid Vectors with Weak Promoters | Enables low, physiologically relevant expression of FP-fusions, reducing overexpression artifacts and background. | pCAGGS (modified) or vectors with weak endogenous promoters. |
| Microscope Calibration Slides | For validating filter set performance, aligning channels, and quantifying bleed-through coefficients. | Argolight Fluorescent Slide (Patterned) |
| Spectral Unmixing Software | Mathematically separates overlapping emission spectra, virtually eliminating bleed-through noise. | Leica LAS X, Zeiss ZEN, or ImageJ plugins like Linear Unmixing. |
Managing Photobleaching and Cellular Toxicity During Long-Term Imaging
Within the broader context of employing GFP fusions to monitor dynamic protein-protein interactions (PPIs), long-term live-cell imaging presents a significant challenge. The very act of observation—repeated excitation with light—induces photobleaching, degrading fluorescent signal, and phototoxicity, which compromises cellular health and perturbs the biological processes under study. This application note details current strategies and protocols to mitigate these effects, ensuring the acquisition of physiologically relevant data over extended time courses.
Core Challenges in Long-Term PPI Imaging
- Photobleaching: Irreversible destruction of the fluorophore's ability to emit light, leading to signal loss. This is critical in PPI studies where quantification of fluorescence (e.g., FRET, co-localization) over time is essential.
- Phototoxicity: Generation of reactive oxygen species (ROS) from fluorophore excitation causes oxidative damage, altering cell physiology, inducing stress responses, and potentially disrupting the very PPIs being monitored.
Table 1: Comparison of Common Fluorescent Proteins for Long-Term Imaging
| Fluorescent Protein | Excitation Peak (nm) | Emission Peak (nm) | Relative Brightness | Photostability (t½, s)* | Maturation Time (min, 37°C) | Notes for PPI Studies |
|---|---|---|---|---|---|---|
| GFP (e.g., EGFP) | 488 | 507 | 1.0 (reference) | 174 | ~90 | Baseline; prone to bleaching in long-term. |
| mNeonGreen | 506 | 517 | 2.5 | 266 | ~60 | Brighter & more photostable; excellent for tagging low-abundance proteins. |
| mCherry | 587 | 610 | 0.47 | 180 | ~90 | Common red FP; moderate photostability. |
| mScarlet | 569 | 594 | 1.5 | 432 | ~15 | Very bright & photostable red FP; fast maturation ideal for kinetics. |
| SiriusGFP | 355 | 424 | 0.24 | >1000 (UV) | ~30 | UV-excitable; minimizes light exposure in visible spectrum. |
| TagRFP-T | 555 | 584 | 0.81 | 441 | ~100 | High photostability; good for red-channel long-term tracking. |
*Photostability half-time under defined illumination conditions. Values are representative and instrument-dependent.
Table 2: Efficacy of Antioxidant & Scavenger Reagents
| Reagent | Mechanism of Action | Typical Working Concentration | Reported Reduction in Phototoxicity | Potential Interference with Biology |
|---|---|---|---|---|
| Ascorbic Acid (Vitamin C) | Direct ROS scavenger | 0.5 - 1.0 mM | Up to 70% | Can affect iron metabolism, be pro-oxidant at high [ ]. |
| Trolox | Vitamin E analog; lipid-soluble antioxidant | 100 - 200 µM | 40-60% | Generally well-tolerated; gold standard for imaging. |
| Pyruvate | Metabolic substrate; scavenges H₂O₂ | 5 - 10 mM | 50-70% | Alters cellular metabolism. |
| Oxyrase | Enzymatic O₂ scavenger | 0.3 - 1.5 U/mL | Significant (O₂ depletion) | Drastically reduces oxygen; can alter hypoxia pathways. |
| Hemoglobin | O₂ binding and ROS scavenging | 0.1 - 0.5 mg/mL | High | Complex to use; may not be suitable for all cells. |
Detailed Protocols
Protocol 4.1: Optimized Media Formulation for Long-Term PPI Imaging
Objective: To prepare imaging media that minimizes phototoxicity while maintaining cell health. Materials: Phenol-red free culture medium, HEPES buffer, Trolox, ascorbic acid, fetal bovine serum (FBS). Procedure:
- Begin with phenol-red free medium appropriate for your cell line.
- Supplement with 25mM HEPES buffer (pH 7.4) to maintain pH without CO₂ control.
- Add 100 µM Trolox (from a 100 mM stock in DMSO or water) and 0.5 mM Sodium L-Ascorbate (from a 100 mM aqueous stock, filter-sterilized).
- Add standard serum (e.g., 10% FBS) if required. Note: Some serum batches contain antioxidants.
- Adjust pH to 7.4 if necessary, filter sterilize (0.22 µm), and store at 4°C protected from light for up to 1 week.
- Warm to 37°C before use. For best results, replace standard medium with this imaging medium 30-60 minutes prior to imaging.
Protocol 4.2: Microscope Configuration for Reduced Photodamage
Objective: To adjust imaging hardware and software parameters for maximal signal-to-noise with minimal light dose. Materials: Inverted epifluorescence or confocal microscope with environmental chamber, high-quantum-efficiency camera, 40x or 60x oil-immersion objective (high NA). Procedure:
- Environmental Control: Stabilize chamber at 37°C and 5% CO₂ (or use HEPES-buffered media) for at least 1 hour before imaging.
- Light Path:
- Use the longest practical wavelength to excite your fluorophore (e.g., use 514 nm for EYFP instead of 488 nm if possible).
- Ensure laser or lamp power is minimized. Start at 0.1-1% laser power for confocal, or use neutral density filters for widefield.
- Detection:
- Camera: Use maximum gain (or EM gain on EMCCD/sCMOS) to allow lower light intensity. Ensure cooling is active to reduce dark noise.
- Confocal: Open the pinhole to 2-3 Airy Units to collect more light, allowing lower laser power.
- Acquisition Settings:
- Use the lowest possible illumination intensity that provides a detectable signal.
- Increase camera exposure time or confocal dwell time to compensate.
- Reduce temporal resolution (increase time interval) to the minimum required for your PPI kinetics.
- Use focus-stabilization systems (e.g., hardware autofocus) to avoid repeated brightfield illumination.
Visualization of Strategies
Mitigation Strategy Workflow for Long-Term PPI Imaging
Mechanism of Photodamage and Key Intervention Points
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Long-Term Live-Cell PPI Imaging
| Item | Example Product/Category | Function & Rationale |
|---|---|---|
| Photostable FPs | mNeonGreen, mScarlet, SiriusGFP | High brightness and resistance to bleaching ensure sustained signal for PPI quantification over hours. |
| Antioxidants | Trolox, Ascorbic Acid (water-soluble) | Scavenge free radicals and ROS generated during imaging, reducing oxidative stress and phototoxicity. |
| Oxygen Scavenging Systems | Oxyrase EC, Glucose Oxidase/Catalase systems | Lower dissolved oxygen to inhibit Type II (¹O₂) phototoxicity pathways. |
| Phenol-Red Free Medium | Gibco FluoroBrite DMEM, other specialty media | Eliminates background autofluorescence and potential photosensitization from phenol red. |
| Environmental Chamber | Okolab, Tokai Hit, PeCon stage-top incubators | Maintains precise temperature, humidity, and CO₂ for cell viability during multi-hour experiments. |
| High-NA Objective Lens | 60x/1.4 NA Oil, 40x/1.3 NA Oil | Collects more emitted photons, allowing lower excitation light intensity for the same signal. |
| Sensitive Camera | sCMOS, EMCCD cameras | High quantum efficiency enables detection of weak signals from dim FPs or low-light conditions. |
| Hardware Autofocus | Nikon Perfect Focus, ZDC2 systems | Maintains focus without exposing cells to additional brightfield illumination, reducing light dose. |
This application note details best practices for the quantitative analysis of fluorescence microscopy images, framed within a thesis investigating protein-protein interactions (PPIs) using GFP-based Förster Resonance Energy Transfer (FRET) and Bimolecular Fluorescence Complementation (BiFC) assays. Reliable quantification is paramount for distinguishing true interaction signals from experimental noise, directly impacting conclusions in drug discovery and basic research.
Key Challenges & Principles
Quantitative fluorescence imaging is prone to variability from sources including uneven illumination, camera noise, photobleaching, background autofluorescence, and cell-to-cell heterogeneity. Data normalization and rigorous image processing are essential to generate comparable, reproducible metrics.
Image Processing Pipeline: Best Practices Protocol
This protocol outlines a standardized workflow for analyzing GFP-FRET data.
Protocol 1: Pre-processing & Background Correction
- Objective: Remove systematic noise and correct for non-specific signal.
- Methodology:
- Flat-field Correction: Capture an image of a uniform fluorescent slide (e.g., fluorescein) or calculate an illumination profile from your dataset. For each raw image pixel, apply:
Corrected = (Raw - Dark) / (Flat - Dark). - Background Subtraction: Define multiple Regions of Interest (ROIs) in cell-free areas of the image. Calculate the mean intensity of these ROIs for each channel. Subtract this mean background value from every pixel in the corresponding channel.
- Smoothing (if required): Apply a mild Gaussian filter (σ=1-2 pixels) only to reduce high-frequency noise, avoiding loss of resolution.
- Flat-field Correction: Capture an image of a uniform fluorescent slide (e.g., fluorescein) or calculate an illumination profile from your dataset. For each raw image pixel, apply:
Protocol 2: Segmentation & ROI Definition
- Objective: Accurately identify and delineate cells or subcellular compartments for measurement.
- Methodology (for cytoplasmic/nuclear analysis):
- Use the channel with the clearest structural signal (e.g., donor GFP or a marker) for segmentation.
- Apply an adaptive thresholding algorithm (e.g., Otsu's method) to create a binary mask.
- Use watershed or seed-based algorithms to separate touching cells.
- For nuclear masking, use a DNA stain (e.g., DAPI) channel. The cytoplasmic mask is defined as the cell mask MINUS the nuclear mask.
- Visually verify all masks and manually correct errors.
Protocol 3: Data Extraction & Normalization
- Objective: Extract biologically meaningful, comparable values.
- Methodology:
- Extraction: For each cell ROI, extract metrics: mean intensity, integrated density, area for all channels (Donor, Acceptor, FRET).
- FRET Correction (for FRET experiments): Calculate corrected FRET (FRETC) using established formulas to remove spectral bleed-through (SBT) and direct acceptor excitation.
where
aandbare SBT coefficients determined from control samples. - Normalization Strategies: Choose based on experimental design (see Table 1).
Table 1: Common Data Normalization Strategies for GFP-PPI Assays
| Normalization Method | Calculation | Purpose | Best For |
|---|---|---|---|
| Ratio-metric (e.g., FRET Efficiency) | FRET_C / Acceptor or FRET_C / Donor |
Corrects for expression level variance & focal plane changes. | FRET, Rationetric biosensors. |
| Background Ratio | (ROI Mean - Background) / Background |
Expresses signal as fold-change over background. | High background scenarios. |
| Z-Score | (X - μ_control) / σ_control |
Expresses data relative to control population statistics. | Comparing treatment effects to a defined control. |
| Total Fluorescence Normalization | ROI Integrated Density / Total Cell Area |
Controls for differences in cell size. | Comparing expression levels across cell populations. |
| Housekeeping Protein | GFP-Fusion Signal / mCherry-Histone Signal |
Corrects for variations in cell number, viability, and transfection efficiency. | Transient transfection experiments. |
Experimental Protocols for Controls
Protocol 4: Determining Spectral Bleed-Through Coefficients
- Express Donor-Only Control: Cells expressing only the GFP-tagged protein.
- Image in Donor and FRET channels using identical acquisition settings as experiment.
- Calculate coefficient
a:a = Mean FRET_channel intensity / Mean Donor_channel intensity. - Repeat with Acceptor-Only control (e.g., mCherry-tagged protein) to calculate coefficient
b(b = FRET / Acceptor).
Protocol 5: Validating a FRET or BiFC Interaction
- Positive Control: Use a known, constitutive heterodimer fusion (e.g., GFP-linker-mCherry).
- Negative Control: Use non-interacting protein pairs or a mutant known to disrupt binding.
- Acquire Images for all samples (test, positive, negative) in the same session with identical settings.
- Process and Normalize all data through the same pipeline.
- Statistical Analysis: Perform one-way ANOVA comparing the normalized FRET ratio (e.g., FRETC/Donor) of test samples against negative and positive controls.
Signaling Pathway & Experimental Workflow
Diagram 1: PPI detection via FRET & quantitative analysis workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Quantitative GFP-PPI Imaging
| Item | Function & Rationale |
|---|---|
| Validated GFP-Fusion Constructs | Ensure the fluorescent tag does not alter protein localization or function. Use low-affinity tags (e.g., ALFA-tag) as an alternative if functionality is impaired. |
| Spectral Bleed-Through Controls | Plasmids for Donor-Only and Acceptor-Only expression are non-negotiable for quantitative FRET. |
| Cell Line with Low Autofluorescence | Use well-characterized lines (e.g., HEK293, HeLa) to minimize background noise. |
| Uniform Fluorescent Reference Slide | Critical for generating a flat-field correction image. |
| Live-Cell Imaging Medium | Phenol-red free, with buffers to maintain pH without CO2. Reduces background and maintains cell health. |
| Validated Positive & Negative Control Plasmids | Essential for calibrating the dynamic range of your assay and validating sensitivity. |
| Automated Microscope with Environmental Control | Enables stable, time-lapse imaging and eliminates temperature/CO2 fluctuations as a variable. |
| Open-Source Analysis Software (e.g., ImageJ/FIJI, CellProfiler) | Provides transparent, customizable algorithms for processing and batch analysis, ensuring reproducibility. |
| High-Quality Objective Lens (60x/63x oil, NA ≥1.4) | Maximizes light collection and spatial resolution, crucial for detecting subtle subcellular changes. |
Beyond Fluorescence: Validating GFP-PPI Data and Comparing Methodologies
Within a thesis investigating GFP-fusion proteins for monitoring protein-protein interactions (PPIs), initial observations from fluorescence imaging require rigorous biochemical validation. Co-immunoprecipitation (Co-IP), Proximity Ligation Assay (PLA), and biochemical fractionation are cornerstone techniques for this purpose. However, each method is susceptible to artifacts, making the inclusion of essential negative and positive controls critical for data integrity and publication. These controls are non-negotiable for researchers and drug development professionals who rely on accurate PPI data for target identification and validation.
The Critical Role of Controls in PPI Validation
Co-Immunoprecipitation (Co-IP) Controls
Co-IP is the gold standard for confirming physical interactions but is prone to false positives from non-specific binding or antibody cross-reactivity.
Essential Controls:
- Negative Control IgG: Use pre-immune serum or an isotype-matched irrelevant antibody to establish background binding levels.
- Negative Cell Line/Tissue: Use lysates from cells lacking the bait protein (e.g., knockout cells) or expressing an irrelevant tagged protein.
- Bait-Only Control: Express the bait (GFP-fusion) without the putative prey partner.
- Prey-Only Control: Express the putative prey without the bait GFP-fusion to detect non-specific binding to the resin or antibody.
- Positive Control: A well-characterized interacting pair, ideally using the same tag/cell system.
Table 1: Quantitative Interpretation of Co-IP Controls
| Control Type | Expected Signal in Prey Detection | Acceptable Outcome | Typical Quantification (Band Intensity vs. Input) |
|---|---|---|---|
| Specific Co-IP | Strong | Prey co-precipitates with bait. | >5-10x over negative IgG control |
| Negative IgG | Weak/Absent | Baseline for non-specific binding. | Set as 1.0 (reference) |
| Bait-KO Lysate | Absent | Confirms antibody specificity. | ≤ 0.5x negative IgG |
| Bait-Only Lysate | Absent | Prey does not bind resin/bead alone. | ≤ 0.5x negative IgG |
| Positive Control | Strong | Validates experimental workflow. | Comparable to literature/prior data |
Proximity Ligation Assay (PLA) Controls
PLA confirms proximal interaction (<40 nm) in situ. Controls validate signal specificity beyond mere co-localization.
Essential Controls:
- Single Primary Antibody Controls: Omit one primary antibody at a time. Should result in >95% reduction in PLA signal.
- Negative Cell Control: Use cells lacking one or both interaction partners.
- Competition Control: Pre-incubate antibody with its blocking peptide.
- Technical Negative: Omit ligase or polymerase from the detection reaction.
- Positive Control: A known interacting pair with validated antibodies.
Table 2: Quantitative PLA Control Outcomes
| Control Type | Expected PLA Signal (# foci/cell) | Acceptable Result |
|---|---|---|
| Full Assay (Test) | Variable, specific | Significant signal above all negatives |
| Omit Primary Ab #1 | ≤ 5% of full assay | Confirms both Abs are required |
| Omit Primary Ab #2 | ≤ 5% of full assay | Confirms both Abs are required |
| Negative Cell Line | ≤ 5% of full assay | Confirms target presence is required |
| Positive Control Pair | High, consistent | Validates reagent and protocol efficacy |
Biochemical Fractionation Controls
Fractionation (e.g., nuclear/cytoplasmic) determines subcellular localization of interactions. Controls ensure fraction purity and validate observed shifts.
Essential Controls:
- Purity Markers: Detect compartment-specific proteins in each fraction (e.g., Lamin B1 for nuclear, GAPDH for cytoplasmic).
- Cross-Contamination Check: The marker for one compartment should be absent/minimal in the other.
- Integrity Control: For membrane-bound organelles, probe for a soluble protein that should not be present (e.g., cytosolic protein in microsomal fraction).
- Total Lysate: Analyze input sample to confirm protein expression.
Table 3: Fractionation Purity Assessment
| Fraction | Target Protein | Compartment Marker | Contamination Marker | Ideal Result |
|---|---|---|---|---|
| Nuclear | Present/Absent | High (Lamin, Histone) | Low (GAPDH, Tubulin) | Marker >>95% in correct fraction |
| Cytosolic | Present/Absent | High (GAPDH, Tubulin) | Low (Lamin, Histone) | Marker >>95% in correct fraction |
Detailed Protocols
Protocol 1: Controlled Co-Immunoprecipitation for GFP-Fusion Proteins
Application: Validating putative interactions identified via GFP-based imaging. Reagents: Anti-GFP nanobody/resin, matched control resin, lysis buffer (25mM Tris pH 7.4, 150mM NaCl, 1% NP-40, protease inhibitors), wash buffer, elution buffer (2x Laemmli buffer).
Method:
- Prepare Lysates: Harvest transfected cells (GFP-bait + putative prey, and all control conditions) in ice-cold lysis buffer. Incubate 30 min on ice, centrifuge at 16,000xg for 15 min.
- Pre-clear: Incubate supernatant with control resin for 30 min at 4°C. Retain input sample.
- Immunoprecipitation: Split pre-cleared lysate. Incubate equal parts with anti-GFP resin and control resin for 2 hours at 4°C.
- Wash: Pellet beads, wash 4x with 1mL wash buffer.
- Elute: Resuspend beads in 40μL 2x Laemmli buffer, boil 5 min.
- Analysis: Run input, unbound, and eluate fractions by SDS-PAGE. Probe for GFP (bait) and candidate prey protein. Quantify band intensity; prey must be enriched in GFP-IP vs. control IP.
Protocol 2: Validated Proximity Ligation Assay (PLA)
Application: Visualizing and quantifying GFP-fusion protein interactions in fixed cells. Reagents: Duolink PLA kit, primary antibodies (anti-GFP and anti-prey), species-specific PLA probes, mounting medium with DAPI.
Method:
- Cell Fixation: Culture cells on chamber slides, transfer, fix with 4% PFA for 15 min, permeabilize with 0.1% Triton X-100.
- Blocking: Incubate with blocking solution for 1h at 37°C.
- Primary Antibodies: Apply anti-GFP and anti-prey antibodies diluted in antibody diluent overnight at 4°C. Include single-antibody controls.
- PLA Probe Incubation: Add species-specific PLUS and MINUS PLA probes, incubate 1h at 37°C.
- Ligation & Amplification: Perform ligation (30 min at 37°C) followed by amplification (100 min at 37°C) per kit instructions.
- Mounting & Imaging: Mount slides with Duolink In Situ Mounting Medium with DAPI. Image using a fluorescence microscope with a 60x objective. Count PLA signals (red foci) per cell in >30 cells.
Protocol 3: Controlled Biochemical Fractionation
Application: Determining if a GFP-fusion protein interaction is compartment-specific. Reagents: Hypotonic lysis buffer (10mM HEPES pH 7.9, 1.5mM MgCl2, 10mM KCl, protease inhibitors), cytoplasmic extraction buffer (above + 0.1% NP-40), nuclear extraction buffer (20mM HEPES pH 7.9, 1.5mM MgCl2, 420mM NaCl, 0.2mM EDTA, 25% glycerol).
Method:
- Harvest Cells: Pellet cells, wash with PBS.
- Cytoplasmic Fraction: Resuspend pellet in hypotonic lysis buffer, incubate on ice 15 min. Add NP-40 to 0.1%, vortex, centrifuge at 12,000xg for 5 min. Supernatant = cytoplasmic fraction.
- Nuclear Fraction: Wash the pellet 2x. Resuspend in nuclear extraction buffer, incubate on ice with vigorous vortexing every 5 min for 30 min. Centrifuge at 16,000xg for 15 min. Supernatant = nuclear fraction.
- Analysis: Run equal percentage volumes of each fraction by SDS-PAGE. Probe for GFP-bait, prey, and purity markers (GAPDH, Lamin B1). Signal should correlate with the compartment-specific marker.
Experimental Workflow & Pathway Diagrams
Workflow for Validating GFP-Based PPI Observations
Validation Pathways for a Putative GFP-Fusion Protein Interaction
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Primary Function in Validation | Key Consideration for GFP-Fusion Studies |
|---|---|---|
| Anti-GFP Nanobody/Antibody | High-affinity capture or detection of GFP-tagged bait protein. | Use for Co-IP and PLA. Prefer nanobodies for milder elution. |
| Magnetic Protein A/G Beads | Solid-phase support for antibody-based Co-IP. | Low non-specific binding essential for clean controls. |
| Duolink PLA Probes & Kits | Generate amplified, detectable signal from proximal (<40nm) antibodies. | Optimal for fixed cells from live-imaging experiments. |
| Compartment-Specific Marker Antibodies | Assess purity in fractionation (e.g., Lamin B1, GAPDH, Cox IV). | Critical controls for localization studies. |
| Protease/Phosphatase Inhibitor Cocktails | Maintain protein integrity and modification state during lysis. | Essential for preserving labile or phosphorylation-dependent PPIs. |
| Validated Positive Control Lysates/Cells | Provide known interaction pair for protocol optimization. | e.g., Lysate from cells expressing GFP and anti-GFP nanobody fusion. |
| Isotype Control IgG | Matched negative control antibody for Co-IP. | Must be same host species, isotype, and conjugation as primary Ab. |
| Cell Line with Target Knockout | Definitive negative control for antibody specificity. | Use CRISPR-generated lines lacking your bait or prey protein. |
Application Notes
Within the broader thesis that GFP-fusion proteins represent a versatile and physiologically relevant platform for monitoring protein-protein interactions (PPIs) in living systems, this analysis compares three pivotal technologies. GFP-based assays, Yeast Two-Hybrid (Y2H), and Surface Plasmon Resonance (SPR) each offer distinct advantages and limitations for different stages of PPI research and drug discovery.
GFP-Based Assays (e.g., FRET, BiFC, FLIM): These live-cell methods leverage genetic fusion of GFP or its variants to proteins of interest. They enable real-time, subcellularly resolved quantification of PPIs under near-physiological conditions. They are ideal for kinetic studies, pathway mapping in relevant cell types, and primary screening in a cellular context. However, they can be susceptible to photobleaching and require careful controls for expression levels and fusion-induced artifacts.
Yeast Two-Hybrid (Y2H): A classic, genetic in vivo system for binary interaction mapping. It is exceptionally high-throughput and cost-effective for screening cDNA libraries against a bait protein, making it unparalleled for de novo discovery of novel interactors. Its main drawbacks are the non-physiological yeast environment (lacking mammalian post-translational modifications) and a significant rate of false positives/negatives.
Surface Plasmon Resonance (SPR): A label-free, in vitro biophysical technique that provides precise kinetic data (association/dissociation rates, equilibrium constants) and stoichiometry. It is the gold standard for validating interactions, characterizing binding affinity, and studying small molecule inhibitors. Its key limitation is the requirement for purified components, removing interactions from their cellular context.
The choice of technique is dictated by the research question: Y2H for discovery, GFP-based assays for cellular validation and dynamics, and SPR for biophysical characterization and drug candidate profiling.
Quantitative Comparison Data
Table 1: Core Methodological Comparison
| Feature | GFP-Based Assays (FRET/BiFC) | Yeast Two-Hybrid (Y2H) | Surface Plasmon Resonance (SPR) |
|---|---|---|---|
| Throughput | Medium (well-plate based) | Very High (library screens) | Low to Medium |
| Environment | Live Cells (Physiological) | In Vivo (Yeast Nucleus) | In Vitro (Purified) |
| Readout | Fluorescence Intensity/Lifetime | Reporter Gene Growth/Color | Refractive Index Shift (RU) |
| Kinetic Data | Yes (Real-time, semi-quantitative) | No (Endpoint) | Yes (Precise ka, kd) |
| Affinity Range | nM to µM (Context-dependent) | Broad (but qualitative) | pM to mM (Direct measurement) |
| Key Artifact | Overexpression, Spectral Bleed-through | Auto-activation, False Positives | Nonspecific Surface Binding |
| Primary Application | Cellular Dynamics, Pathway Mapping | Interactome Discovery | Mechanistic & Drug Binding Studies |
Table 2: Typical Experimental Metrics
| Metric | GFP-FRET | Yeast Two-Hybrid | SPR (Biacore) |
|---|---|---|---|
| Assay Development Time | 2-4 weeks (clone generation) | 1-2 weeks | 1-3 weeks (protein purification) |
| Data Acquisition Time | Seconds to minutes per sample | 3-5 days for colony growth | Minutes per analyte injection |
| Sample Consumption | Low (ng of DNA) | Low | Moderate (µg of purified protein) |
| Typical Kd Measurement | Semi-quantitative (~% FRET efficiency) | Not directly measurable | Quantitative (e.g., 10 nM ± 2 nM) |
| False Positive Rate | Low with controls | Can be >50% in screens | Very Low with proper design |
Experimental Protocols
Protocol 1: GFP-Based FRET by Acceptor Photobleaching
Objective: To confirm and quantify a PPI in live mammalian cells by measuring an increase in donor fluorescence after selective bleaching of the acceptor. Key Controls: Cells expressing donor-only and acceptor-only constructs.
- Construct Generation: Clone genes of interest (POIs) into vectors encoding donor (eGFP) and acceptor (mCherry) fluorescent proteins. Create N- or C-terminal fusions as appropriate.
- Cell Transfection: Seed HEK293T cells in an imaging-compatible 8-well chamber slide. Transfect with a 1:1 ratio of donor and acceptor fusion construct DNA using a transfection reagent. Include control wells.
- Imaging Setup (24-48h post-transfection): Use a confocal microscope with appropriate lasers (488 nm for eGFP, 561 nm for mCherry). Set up sequential scanning to minimize bleed-through. Define a region of interest (ROI) in the cell cytoplasm/nucleus.
- Pre-bleach Image Acquisition: Capture donor (eGFP) and acceptor (mCherry) channel images of the ROI.
- Acceptor Photobleaching: Using high-intensity 561 nm laser light, bleach the mCherry signal in the selected ROI until fluorescence is reduced by >80%.
- Post-bleach Image Acquisition: Immediately re-capture the donor (eGFP) channel image using the same settings as in step 4.
- Data Analysis: Calculate FRET efficiency (E) for each cell using the formula:
E = (I_post - I_pre) / I_post, whereIis the donor fluorescence intensity in the bleached ROI. A significant increase in donor fluorescence post-bleach indicates FRET and thus proximity (<10 nm) of the POIs.
Protocol 2: Standard Yeast Two-Hybrid Screening
Objective: To identify novel protein partners (Prey) for a protein of interest (Bait).
- Bait Construct Creation: Clone the cDNA of the Bait protein into the pGBKT7 (or similar) vector, creating a fusion with the GAL4 DNA-Binding Domain (DBD). Transform into yeast strain Y2HGold and plate on SD/-Trp to select for bait plasmid.
- Bait Validation: Check for auto-activation by plating bait strain on high-stringency SD/-Trp/-His/-Ade plates. A bait that activates reporter genes alone is unsuitable.
- Mating Screen: Mate the bait strain with a pre-transformed library of prey proteins (fused to GAL4 Activation Domain in pGADT7) in yeast strain Y187. Perform mating in 2x YPDA broth overnight.
- Diploid Selection: Plate the mating mixture on high-stringency SD/-Trp/-Leu/-His/-Ade + X-α-Gal plates. Incubate at 30°C for 3-7 days.
- Colony Analysis: Pick blue, growing colonies. Isolate the prey plasmid DNA and sequence to identify the interacting protein candidate.
- Confirmation: Re-transform isolated prey plasmid with the original bait plasmid into fresh yeast to confirm the interaction.
Protocol 3: SPR Analysis of a Protein-Small Molecule Interaction
Objective: To determine the binding kinetics (ka, kd) and affinity (KD) of a small molecule inhibitor for its target protein.
- Surface Preparation: Using a Biacore T200 system, immobilize the recombinant target protein onto a CM5 sensor chip via amine coupling to achieve a response of ~5000 RU.
- Running Buffer Preparation: Prepare HBS-EP+ buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% v/v Surfactant P20, pH 7.4). Use this for dilution and as running buffer.
- Analyte Series: Prepare a 2-fold dilution series of the small molecule analyte (e.g., from 100 nM to 1.56 nM) in running buffer.
- Binding Experiment: Use a multi-cycle kinetics method. Inject each analyte concentration over the protein surface and a reference flow cell for 120 s (association phase), followed by a 300 s dissociation phase with buffer flow. Regenerate the surface with a short pulse of regeneration buffer (e.g., 10 mM glycine pH 2.0).
- Data Processing: Subtract the reference flow cell signal. Fit the resulting sensorgrams globally to a 1:1 binding model using the Biacore Evaluation Software.
- Analysis: The software will report the association rate constant (ka, M⁻¹s⁻¹), dissociation rate constant (kd, s⁻¹), and the equilibrium dissociation constant (KD = kd/ka, M).
Diagrams
Title: FRET by Acceptor Photobleaching Protocol Flow
Title: Decision Tree for PPI Method Selection
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Featured Protocols
| Reagent/Material | Function/Application | Key Consideration |
|---|---|---|
| pEGFP-N1 & mCherry-C1 Vectors | Backbones for creating C-terminal and N-terminal GFP/RFP fusion proteins, respectively. | Choose linker sequence between POI and fluorophore to minimize steric interference. |
| HEK293T Cells | Highly transferable mammalian cell line for transient expression of GFP-fusion constructs. | Maintain low passage number for consistent transfection efficiency. |
| Polyethylenimine (PEI) | Cationic polymer transfection reagent for efficient DNA delivery into HEK293T cells. | Optimize DNA:PEI ratio for each construct to balance expression and toxicity. |
| SD/-Trp/-Leu/-His/-Ade Medium | Selective yeast dropout medium for Y2H diploid selection. Stringency prevents false positives. | Supplement with X-α-Gal for colorimetric (blue/white) reporter selection. |
| Y2HGold & Y187 Yeast Strains | Genetically engineered S. cerevisiae with multiple reporter genes (AUR1-C, ADE2, HIS3, MEL1) for sensitive Y2H. | Check mating type (MATa vs MATα) for appropriate mating. |
| CM5 Sensor Chip (Biacore) | Gold surface with carboxymethylated dextran matrix for covalent immobilization of proteins via amine coupling. | The workhorse chip for most protein-ligand SPR studies. |
| HBS-EP+ Buffer | Standard SPR running buffer. Provides stable pH and ionic strength, and contains surfactant to minimize nonspecific binding. | Must be degassed to prevent air bubbles in the microfluidic system. |
| Series S Sensor Chip NTA | For capturing His-tagged proteins via nickel chelation. Allows for regeneration and reuse of the chip with different ligands. | Ideal for screening where multiple baits are used. Requires NiCl2 activation. |
Within the broader thesis of utilizing GFP fusions for monitoring protein-protein interactions (PPIs), this document provides a critical assessment of three fundamental assay characteristics: sensitivity, throughput, and physiological relevance. GFP-based PPI assays, such as Förster Resonance Energy Transfer (FRET), Bimolecular Fluorescence Complementation (BiFC), and fluorescence correlation spectroscopy (FCS), are indispensable tools. However, their utility in drug discovery and basic research depends on a clear understanding of their performance trade-offs. This application note details experimental protocols, comparative data, and essential reagents to guide researchers in selecting and optimizing the appropriate GFP-based strategy.
Core Assay Comparison
The following table summarizes the quantitative performance characteristics of three primary GFP-based PPI techniques.
Table 1: Comparison of Key GFP-Based PPI Assay Modalities
| Assay Characteristic | FRET (e.g., GFP-RFP Pair) | Bimolecular Fluorescence Complementation (BiFC) | Fluorescence Fluctuation Analysis (e.g., FCCS) |
|---|---|---|---|
| Sensitivity (Detection Limit) | Moderate. Requires ~10-100 nM affinity. High background from donor bleed-through and direct acceptor excitation. | High for stable interactions. Can detect weak/transient interactions due to irreversible complementation. | Very High. Can detect interactions at single-molecule concentrations (<1 nM). |
| Throughput | High. Compatible with microplate readers for endpoint or kinetic live-cell assays. | Moderate to Low. Slow maturation of complemented fluorophore (minutes to hours) limits kinetic studies. | Low. Requires specialized confocal microscopes; data acquisition and analysis are time-intensive. |
| Physiological Relevance | High when performed in live cells. Reports on proximity (<10 nm) in real-time. Can be confounded by overexpression. | Caution Required. Irreversible complex can trap transient interactions, potentially creating artifacts. | Very High. Performed at endogenous expression levels (e.g., via CRISPR knock-in). Measures diffusion and co-diffusion in native membrane/organelle environments. |
| Key Quantitative Metric | FRET Efficiency (E%), typically 5-30% for positive interactions. | Fluorescence Intensity (counts/sec) of complemented signal vs. controls. | Co-diffusion Correlation (Cross-correlation amplitude, Gcc(0)). |
| Typical Assay Time | Seconds to minutes for kinetic monitoring. | Hours (due to fluorophore maturation). | Minutes to hours per sample. |
Detailed Experimental Protocols
Protocol 1: Live-Cell FRET Acceptor Photobleaching Assay
This protocol measures FRET efficiency by quantifying the increase in donor fluorescence after bleaching the acceptor, confirming direct molecular proximity.
I. Materials & Reagents
- Cells (e.g., HEK293T, HeLa) expressing donor (GFP) and acceptor (RFP/mCherry) fusion proteins.
- Microscope with controllable laser lines for 488 nm (GFP excitation) and 561 nm (RFP excitation), and a bleaching function at 561 nm.
- Imaging chamber with CO₂ and temperature control.
- Phenol-red free imaging medium.
II. Procedure
- Cell Preparation: Plate cells on glass-bottom dishes. Transfect with constructs for Donor-GFP and Acceptor-RFP fusion proteins. Incubate for 24-48 hours.
- Image Acquisition Setup: Using a confocal microscope, set up sequential scanning to avoid cross-talk.
- Channel 1 (Donor): Excitation 488 nm, Emission 500-550 nm.
- Channel 2 (Acceptor): Excitation 561 nm, Emission 570-620 nm.
- Pre-bleach Imaging: Identify a region of interest (ROI) expressing both fluorophores. Acquire 3-5 pre-bleach images for both channels.
- Acceptor Photobleaching: Select the same ROI for bleaching. Bleach the acceptor fluorophore using the 561 nm laser at 100% intensity for 30-60 iterations.
- Post-bleach Imaging: Immediately re-acquire images in both channels using the same settings as in step 3.
- Data Analysis:
- Measure the mean fluorescence intensity in the donor channel (GFP) within the ROI before (IDpre) and after (IDpost) bleaching.
- Calculate FRET Efficiency: E = (IDpost – IDpre) / IDpost.
- A significant increase in donor fluorescence after acceptor bleaching indicates positive FRET.
Protocol 2: Bimolecular Fluorescence Complementation (BiFC) Assay
This protocol detects protein interactions by visualizing the reconstitution of a fluorescent protein from two non-fluorescent fragments.
I. Materials & Reagents
- BiFC plasmid vectors (e.g., pBiFC-VN173 and pBiFC-VC155 for Venus-YFP).
- Appropriate cell line.
- Standard cell culture and transfection reagents.
- Fluorescence microscope or plate reader with appropriate filters (e.g., YFP: Ex 515 nm, Em 528 nm).
II. Procedure
- Construct Cloning: Clone your proteins of interest (POI) into the BiFC vectors, generating fusions with the N-terminal (VN) and C-terminal (VC) fragments of the split fluorescent protein.
- Transfection: Co-transfect cells with the following plasmid pairs:
- Test: POI-A-VN + POI-B-VC
- Negative Control 1: POI-A-VN + VC-only (or unrelated protein-VC)
- Negative Control 2: VN-only + POI-B-VC
- Positive Control: Known interacting pair fused to VN/VC.
- Expression & Maturation: Incubate transfected cells for 24-48 hours to allow for protein expression and the irreversible maturation of the complemented fluorophore.
- Imaging/Quantification:
- Microscopy: Image live or fixed cells. Reconstituted fluorescence is typically localized to the subcellular compartment of the interaction.
- Plate Reader: For higher throughput, lysate cells and measure fluorescence intensity in a microplate. Normalize fluorescence to total protein concentration or a co-transfected control (e.g., Renilla luciferase).
- Analysis: Signal in the test sample must be significantly higher than in both negative controls to claim a specific interaction.
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for GFP-Based PPI Studies
| Reagent / Material | Function & Relevance |
|---|---|
| FRET-Calibrated FP Pairs (e.g., mTurquoise2-sfGFP, mNeonGreen-mRuby3) | Optimized donor-acceptor pairs with high quantum yield, photostability, and minimal spectral bleed-through for sensitive, quantitative FRET. |
| CRISPR/Cas9 Knock-in Cell Lines | Enables tagging of endogenous proteins with GFP/RFP at native loci, eliminating overexpression artifacts and enhancing physiological relevance. |
| Phenol-Red Free / Live-Cell Imaging Medium | Reduces background autofluorescence for sensitive detection of fluorescent signals in live cells over time. |
| Acceptor Photobleaching / FLIM-FRET Module | Specialized microscope hardware/software essential for performing and analyzing acceptor photobleaching or Fluorescence Lifetime Imaging (FLIM)-FRET experiments. |
| Split FP Vectors (e.g., Venus-YFP [VN/VC], sfCherry2 [VH/VL]) | Validated, codon-optimized plasmids for BiFC assays, offering varying maturation speeds and spectral properties. |
| Fluorescence Fluctuation Spectroscopy Software (e.g., SimFCS, FoCuS-point) | Specialized analysis software required to calculate autocorrelation and cross-correlation curves from FCS and FCCS data. |
Visualizations
Decision Workflow for GFP-Based PPI Assay Selection
Mechanisms of FRET Acceptor Bleaching and BiFC
Within the broader thesis on GFP-fusion proteins for monitoring protein-protein interactions (PPIs), the critical challenge remains establishing that observed fluorescence signals—whether from FRET, fluorescence polarization, or co-localization—genuinely reflect biologically meaningful functional outcomes. This application note details protocols and frameworks for correlating quantitative fluorescence data with downstream cellular or organismal phenotypes, establishing the gold standard for validating PPI assays in drug discovery and basic research.
Key Quantitative Correlations: Validation Benchmarks
The following table summarizes established quantitative relationships between fluorescence-based PPI metrics and functional outcomes, essential for assay validation.
Table 1: Fluorescence-Function Correlation Benchmarks
| Fluorescence Metric (GFP-based) | Correlated Functional Outcome | Assay Type | Typical Correlation Coefficient (R²) | Key Validating Experiment |
|---|---|---|---|---|
| FRET Efficiency (E%) | Kinase Activation (e.g., ERK) | FRET Biosensor | 0.85 - 0.95 | Phospho-specific immunoblotting |
| Fluorescence Co-localization (Manders' Coefficient) | Vesicle Trafficking / Fusion | Confocal Microscopy | 0.70 - 0.90 | Electron microscopy; cargo release assay |
| Bimolecular Fluorescence Complementation (BiFC) Signal Intensity | Transcriptional Activation | BiFC + Reporter Gene | 0.75 - 0.85 | Luciferase reporter assay; qPCR of target genes |
| Fluorescence Polarization (mP) Shift | Ligand-Receptor Binding (Inhibition) | Competitive Binding FP | 0.90 - 0.98 | Radioligand binding assay (Ki determination) |
| Protein Fragment Complementation (e.g., GFP-GFP) Luminescence | Pathway-Specific Apoptosis | PCA (Protein Complementation) | 0.80 - 0.92 | Caspase-3/7 activity assay; Annexin V flow cytometry |
Detailed Protocols
Protocol 1: Validating FRET Biosensor Data with Phospho-Protein Immunoblotting
This protocol correlates FRET efficiency changes from a GFP-RFP biosensor (e.g., for ERK activity) with standard western blot analysis.
- Cell Preparation & Transfection: Seed HEK293T cells in a 6-well plate for blotting and a 35mm glass-bottom dish for imaging. Co-transfect with the FRET biosensor (e.g., EKAR) and a relevant receptor plasmid using a polyethylenimine (PEI) method.
- Stimulation & Parallel Sampling: Starve cells in serum-free medium for 24h. For each time point (0, 5, 15, 30 min), prepare one well for lysis and one dish for imaging. Stimulate with ligand (e.g., 100 ng/mL EGF).
- FRET Imaging & Analysis: Image using a confocal microscope with 405 nm excitation. Collect emissions at 460±30 nm (CFP) and 535±25 nm (FRET). Calculate FRET ratio (FRET/CFP) per cell using image analysis software (e.g., ImageJ/FIJI).
- Cell Lysis & Immunoblotting: Immediately lyse parallel wells in RIPA buffer with protease/phosphatase inhibitors. Run 20 µg protein on SDS-PAGE, transfer to PVDF, and blot for phospho-ERK (p-p44/42) and total ERK.
- Correlation Analysis: Normalize both FRET ratio and blot band density (ImageJ) to time zero. Plot normalized FRET ratio vs. normalized pERK/ERK ratio for each time point. Perform linear regression to calculate R².
Protocol 2: Correlating BiFC Interaction with Transcriptional Reporter Activity
This protocol validates that a observed PPI via Bimolecular Fluorescence Complementation (BiFC) directly influences downstream gene expression.
- Construct Design: Clone proteins of interest (POIs) as fusions to split GFP fragments (e.g., GFP[1-157] and GFP[158-238]). Clone a luciferase reporter gene under the control of a promoter known to be regulated by the POI interaction.
- Triple Transfection: In a 96-well plate (white wall, clear bottom), co-transfect cells with three plasmids: BiFC-POI-A, BiFC-POI-B, and the firefly luciferase reporter. Include controls (e.g., empty BiFC vectors). Use a renilla luciferase plasmid for normalization.
- Dual Measurement at 48h:
- Fluorescence: Read GFP fluorescence (Ex ~485 nm, Em ~535 nm) to quantify BiFC complex formation.
- Luminescence: Lyse cells per manufacturer protocol (Dual-Glo, Promega). Sequentially measure firefly and renilla luminescence.
- Data Correlation: Calculate normalized reporter activity (Firefly/Renilla). Plot this value against the corresponding well's GFP fluorescence intensity (background subtracted). A strong positive correlation across experimental conditions (e.g., dose-response to an activator) validates the functional link.
Visualizing Correlation Workflows and Pathways
Diagram 1: Gold Standard Validation Workflow
Diagram 2: FRET Reports on a Functional Signaling Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Fluorescence-Function Correlation Studies
| Reagent / Solution | Function in Correlation Studies | Example Product / Note |
|---|---|---|
| Genetically Encoded FRET Biosensors | Provide real-time, rationetric readout of kinase activity or second messengers (e.g., Ca²⁺) in live cells. | EKAR (ERK), AKAR (PKA), Cameleon (Ca²⁺). |
| Split GFP/YFP/Venus Systems (BiFC/PCA) | Enable visualization of weak or transient PPIs by irreversible fluorescent complementation. | Venus [1-158/159-238]; use low temps to reduce false positives. |
| HTRF (Homogeneous Time-Resolved FRET) Kits | Validate cellular PPIs in a plate-reader format with high signal-to-noise; gold standard for biochemical confirmation. | Cisbio PPI kits; uses Eu³⁺ cryptate donor. |
| Phospho-Specific Antibody Panels | Directly measure functional pathway activation downstream of a PPI for correlation with fluorescence data. | CST (Cell Signaling Technology) Phospho-MAPK Array. |
| Live-Cell Compatible Agonists/Antagonists | Modulate PPIs dynamically in fluorescence assays to generate dose-response data for correlation. | Tet System-approved small molecules; iper agonists. |
| Dual-Luciferase Reporter Assay Systems | Quantitate transcriptional functional outcome in parallel with fluorescence imaging from the same sample. | Promega Dual-Glo; allows sequential measurement. |
| Automated Image Analysis Software | Extract robust, high-content quantitative data (ratios, co-localization) from fluorescence images. | ImageJ/FIJI, CellProfiler, MetaMorph. |
| Statistical Analysis Software | Perform regression analysis (linear, non-linear) to calculate correlation coefficients (R², Pearson's r). | GraphPad Prism, R (with ggplot2). |
Within the broader thesis that GFP fusions serve as a central platform for monitoring dynamic protein-protein interactions (PPIs), a critical limitation persists: standard fluorescence imaging confirms co-localization but cannot prove direct physical interaction or identify novel binding partners. Emerging hybrid approaches that integrate GFP imaging with either proximity ligation assays (PLA) or mass spectrometry (MS) directly address this gap. These methods leverage the GFP tag not just for visualization, but as a genetically encoded handle for in situ validation or proteomic discovery, transforming a ubiquitous tool into a gateway for high-specificity interaction analysis.
Application Notes
1. GFP-PLA (Proximity Ligation Assay) for In Situ Validation This approach, often called in situ PLA, uses a primary antibody against GFP and another against a putative interaction partner. If the two proteins are within ~40 nm, oligonucleotide-conjugated secondary antibodies (PLA probes) enable rolling-circle amplification, generating a fluorescent signal detectable as a distinct punctum. This confirms direct or proximate interaction with spatial resolution in fixed cells.
- Key Advantage: Moves beyond co-localization to validate direct PPIs in their native cellular context with single-interaction sensitivity.
- Primary Application: Validating hypothesized interactions from initial GFP-based screens (e.g., FRET, co-localization) in relevant cell models or tissue sections.
2. GFP Affinity Purification / Immunoprecipitation coupled with Mass Spectrometry (GFP-AP/MS) Here, the GFP fusion protein is used as bait for affinity capture under near-physiological conditions. The GFP-tagged protein and its endogenous interactors are purified using anti-GFP nanobodies or antibodies immobilized on beads, followed by identification via liquid chromatography-tandem MS (LC-MS/MS).
- Key Advantage: Unbiased discovery of novel interaction partners and complexes in a single experiment, without requiring antibodies for candidate proteins.
- Primary Application: De novo mapping of interaction networks for a protein of interest, crucial for pathway elucidation and drug target identification.
Quantitative Comparison of Hybrid Approaches Table 1: Comparative Analysis of GFP-Based Hybrid Interaction Methods
| Parameter | GFP-PLA (Validation) | GFP-AP/MS (Discovery) |
|---|---|---|
| Primary Goal | Spatial validation of suspected PPIs | Unbiased identification of novel interactors |
| Throughput | Medium (multiplexable) | Low to Medium |
| Spatial Context | Preserved (in situ, fixed cells/tissue) | Lost (lysate-based) |
| Interaction Proximity | ≤ 40 nm (direct/indirect) | Direct and stable in lysate buffer |
| Typical Output | Number of puncta per cell (counts) | List of proteins with enrichment scores |
| Key Quantitative Readout | ~15-40 puncta/cell for positive interaction vs. 0-2 puncta/cell for negative control. | Fold-change (e.g., >5x) and significance (p-value < 0.05) vs. control purifications. Commonly uses SAINT or CompPASS scores. |
| Sensitivity | Single-molecule detection possible | Requires sufficient material for MS detection (fmol-pmol) |
| Best Suited For | Translational research, diagnostic pathology, pre-clinical target validation | Early-stage discovery, systems biology, complex characterization |
Detailed Experimental Protocols
Protocol A: GFP-Proximity Ligation Assay (GFP-PLA) for PPI Validation
Objective: To validate the direct interaction between a GFP-tagged protein (bait) and an endogenous protein (prey) in fixed HeLa cells.
Research Reagent Solutions & Essential Materials
- Cell Line: HeLa cells expressing GFP-tagged protein of interest.
- Antibodies: Mouse anti-GFP primary antibody, rabbit antibody against endogenous target protein.
- PLA Kit: Duolink In Situ PLA Kit (Sigma-Aldrich, with PLUS and MINUS PLA probes).
- Fixative: 4% Paraformaldehyde (PFA) in PBS.
- Permeabilization Buffer: 0.5% Triton X-100 in PBS.
- Blocking Solution: Duolink Blocking Solution.
- Mounting Medium with DAPI: For nucleus staining.
Methodology:
- Cell Culture & Fixation: Seed cells on chambered coverslips. At 70-80% confluency, fix with 4% PFA for 15 min at RT. Permeabilize with 0.5% Triton X-100 for 10 min.
- Blocking: Incubate with Duolink Blocking Solution in a pre-heated humidity chamber for 60 min at 37°C.
- Primary Antibody Incubation: Dilute mouse anti-GFP and rabbit anti-target antibodies in Duolink Antibody Diluent. Apply to samples and incubate overnight at 4°C in a humidity chamber.
- PLA Probe Incubation: Wash cells 3x with Wash Buffer A (5 min per wash). Apply the oligonucleotide-conjugated PLA probes (anti-mouse PLUS, anti-rabbit MINUS) diluted in Antibody Diluent. Incubate for 60 min at 37°C.
- Ligation: Wash 3x with Buffer A. Add the Ligation-Ligase solution. Incubate for 30 min at 37°C.
- Amplification: Wash 3x with Buffer A. Add the Amplification-Polymerase solution containing fluorescently labeled nucleotides. Incubate for 100 min at 37°C in the dark.
- Detection: Wash 2x with Buffer B, then 1x with 0.01x Buffer B. Let slides dry and mount with Duolink In Situ Mounting Medium with DAPI.
- Imaging & Analysis: Image using a fluorescence microscope with appropriate filters for the PLA fluorophore (e.g., Cy3/Alexa 594) and DAPI. Quantify the number of discrete PLA puncta per cell using image analysis software (e.g., ImageJ/FIJI).
Protocol B: GFP Affinity Purification for Mass Spectrometry (GFP-AP/MS)
Objective: To identify proteins that interact with a GFP-tagged bait protein in HEK293T cells.
Research Reagent Solutions & Essential Materials
- Cell Line: HEK293T cells expressing GFP-tagged bait or GFP-alone control.
- Lysis Buffer: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1x protease/phosphatase inhibitors.
- GFP-Trap Agarose: Magnetic beads coupled to a high-affinity anti-GFP nanobody (ChromoTek).
- Wash Buffer: 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.05% NP-40.
- Elution Buffer: 2x Laemmli sample buffer, or mild elution with 0.2 M Glycine pH 2.5.
- Mass Spectrometry Facility: Access to LC-MS/MS and bioinformatics analysis.
Methodology:
- Cell Lysis: Harvest ~1-2 x 10^7 cells per condition (GFP-bait and GFP-only control). Lyse cells in 1 mL ice-cold Lysis Buffer for 30 min with rotation at 4°C. Clarify lysate by centrifugation at 20,000 x g for 15 min at 4°C.
- Affinity Capture: Pre-clear lysate with protein A/G beads for 30 min. Incubate the supernatant with 25 µL of equilibrated GFP-Trap agarose beads for 2 hours at 4°C with rotation.
- Stringent Washes: Pellet beads magnetically. Wash sequentially: 3x with 1 mL Wash Buffer, 1x with 1 mL of Wash Buffer containing 500 mM NaCl (high-salt wash), and 1x with 1 mL Wash Buffer.
- On-Bead Digestion (Preferred for MS): Resuspend beads in 50 µL of 50 mM Ammonium Bicarbonate. Add 1 µg of trypsin/Lys-C mix and incubate overnight at 37°C. Acidify peptides with formic acid and desalt using C18 StageTips.
- LC-MS/MS Analysis: Analyze peptides by nanoflow LC-MS/MS. Use a 120-min gradient for peptide separation.
- Data Processing & Analysis: Identify proteins using search engines (e.g., MaxQuant, Proteome Discoverer) against a human UniProt database. Compare GFP-bait samples to GFP-only controls using statistical tools (e.g., Significance Analysis of INTeractome - SAINT) to assign probability scores (SAINT score > 0.8) and fold-changes to identified interactors.
Visualizations
GFP-PLA Workflow for PPI Validation
GFP-AP/MS Workflow for Interactor Discovery
Thesis Context: Bridging the GFP PPI Gap
Conclusion
GFP fusion proteins have fundamentally transformed our ability to visualize and quantify protein-protein interactions within the native context of the living cell, offering unparalleled temporal and spatial resolution. This guide has underscored that successful application hinges on a solid foundational understanding, meticulous experimental design, rigorous troubleshooting, and essential orthogonal validation. While techniques like FRET and BiFC provide powerful dynamic data, they are most impactful when integrated into a broader validation framework. The future of GFP-based PPI monitoring lies in the development of brighter, more photostable fluorophores, advanced computational analysis tools for complex interaction networks, and the creation of genetically encoded biosensors for specific disease pathways. For drug development, these evolving tools promise to accelerate target identification, mechanism-of-action studies, and high-content screening, bridging the gap between cellular biochemistry and therapeutic discovery. Mastering these techniques is therefore crucial for advancing both basic molecular biology and translational clinical research.