GFP vs. Bilirubin-Binding Proteins: A Researcher's Guide to Choosing Fluorescent Tags
This article provides a comprehensive comparison between Green Fluorescent Protein (GFP) and bilirubin-inducible fluorescent proteins (FPs) like UnaG, offering biomedical researchers and drug development professionals a structured framework for selecting...
GFP vs. Bilirubin-Binding Proteins: A Researcher's Guide to Choosing Fluorescent Tags
Abstract
This article provides a comprehensive comparison between Green Fluorescent Protein (GFP) and bilirubin-inducible fluorescent proteins (FPs) like UnaG, offering biomedical researchers and drug development professionals a structured framework for selecting the optimal tag. We explore the foundational biology, from GFP's intrinsic β-barrel structure to the fatty acid binding protein architecture of bilirubin-binding FPs. The scope extends to methodological applications in live-cell imaging, biosensors, and therapeutic cell tracking, alongside troubleshooting for common challenges like anaerobic conditions, acidic environments, and autofluorescence. A direct, evidence-based comparison validates the performance of each system, empowering scientists to make informed decisions for their specific experimental or clinical contexts.
Illuminating the Core: Structural and Mechanistic Foundations of Fluorescent Proteins
The discovery and development of the Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria revolutionized molecular and cellular biology, providing researchers with a genetically encodable fluorescent marker that functions without exogenous cofactors [1]. The iconic GFP structure—an 11-stranded β-barrel surrounding a central chromophore—has become a paradigm for understanding how protein architecture can confer remarkable fluorescence properties [2] [3]. This scaffold protects the chromophore from non-radiative decay, allowing efficient fluorescence emission [4]. Within this structural paradigm, a new class of fluorescent proteins has emerged that leverages similar β-barrel scaffolds but operates through fundamentally different mechanisms. Bilirubin-inducible fluorescent proteins, notably UnaG discovered in the Japanese eel Anguilla japonica, represent a significant divergence from the GFP model, utilizing ligand binding rather than autocatalytic chromophore formation to generate fluorescence [5] [6] [7]. This comparative analysis examines the structural foundations, photophysical properties, and experimental applications of these distinct yet structurally related fluorescent protein families, providing researchers with objective data to inform their selection for specific investigative contexts.
Structural Foundations and Fluorescence Mechanisms
The GFP Family: Intrinsic Chromophore Formation
The GFP β-barrel scaffold encloses a chromophore that forms autocatalytically from three consecutive amino acids (X65-Tyr66-Gly67) within the polypeptide chain [2] [1]. This post-translational modification involves cyclization, dehydration, and oxidation reactions that create an extended π-conjugated system responsible for light absorption and emission [3] [1]. The mature chromophore, 4-(4-hydroxybenzylidene)-1,2-dimethyl-imidazolinone (p-HOBDI), is deeply embedded within the β-barrel, where it is shielded from the aqueous environment and its rotational freedom is restricted, thereby enabling high fluorescence quantum yield [4]. A critical limitation of conventional GFP-like proteins is their absolute requirement for molecular oxygen to complete the chromophore maturation process, which precludes their use in anaerobic systems [8].
UnaG and Bilirubin-Binding Proteins: Ligand-Induced Fluorescence
UnaG belongs to the fatty acid-binding protein (FABP) family and features a β-barrel structure that transiently binds bilirubin (BR) as an exogenous fluorophore [5] [6] [7]. Unlike GFP, UnaG in its apo state (apoUnaG) is non-fluorescent; fluorescence emerges only upon formation of the holoUnaG complex with bilirubin [5] [7]. The UnaG-bilirubin interaction is characterized by high specificity, with the ligand protected from solvent within the protein barrel by a conserved glycine-proline-proline (GPP) motif [6]. This ligand-binding mechanism operates independently of oxygen, enabling fluorescence applications in anaerobic environments where GFP cannot function [8]. Recent research has revealed that holoUnaG exists in two distinct fluorescence states (holoUnaG1 and holoUnaG2) that reach equilibrium through a reversible intramolecular reaction, with the brighter holoUnaG2 state exhibiting approximately fourfold greater molecular brightness [5].
Table 1: Fundamental Structural and Functional Characteristics
| Characteristic | GFP Family | UnaG/Bilirubin-Binding FPs |
|---|---|---|
| Structural Scaffold | 11-stranded β-barrel [2] [3] | β-barrel typical of FABP family [5] [6] |
| Chromophore Origin | Autocatalytic from internal tripeptide [2] [1] | Exogenous ligand (bilirubin) [5] [7] |
| Maturation Requirement | Oxygen-dependent oxidation [8] | Oxygen-independent binding [8] |
| Apo-State Fluorescence | Non-fluorescent until matured | Non-fluorescent without ligand [5] [7] |
| Key Conserved Motifs | Ser65-Tyr66-Gly67 (or variants) [2] | Glycine-Proline-Proline (GPP) loop [6] |
| Maturation Time | Minutes to hours [9] | Instantaneous upon ligand binding [5] |
Comparative Photophysical Properties
The practical utility of fluorescent proteins in research depends critically on their photophysical characteristics, which determine their brightness, spectral profile, and stability under experimental conditions. Quantitative comparison of these parameters enables researchers to match protein properties to application requirements.
Table 2: Photophysical Properties and Performance Metrics
| Parameter | GFP (EGFP) | UnaG | Experimental Context |
|---|---|---|---|
| Excitation Maximum (nm) | ~488 [9] | 496-498 [5] [6] | In vitro spectroscopic measurement |
| Emission Maximum (nm) | ~507 [9] | 527-532 [5] [6] | In vitro spectroscopic measurement |
| Quantum Yield (%) | ~50-60 [5] | ~50 [5] | Comparison to known standards |
| Maturation Rate | Minutes to hours [9] | Instantaneous [5] | Time from synthesis to fluorescence |
| Brightness Ratio (States) | N/A | 1:3.9 (holoUnaG1:holoUnaG2) [5] | Photon counting statistics |
| Oxygen Requirement | Essential [8] | None [8] | Anaerobic bacterial imaging |
| Thermal Stability | High (denatures ~65-70°C) [3] | Moderate (mutants alter stability) [7] | Circular dichroism melting curves |
The data reveal that UnaG achieves quantum efficiency comparable to enhanced GFP (approximately 50%) despite their different fluorescence mechanisms [5]. UnaG's instantaneous activation upon bilirubin binding provides a significant advantage for time-sensitive applications, unlike GFP's slower maturation process. The recently discovered two-state system in UnaG adds complexity to its photophysical behavior, with the equilibrium favoring the brighter holoUnaG2 state (6:4 population ratio) [5]. From a practical standpoint, UnaG's oxygen independence enables applications in anaerobic environments that are inaccessible to GFP [8].
Experimental Applications and Methodologies
GFP-Based Experimental Workflows
GFP and its engineered variants have enabled diverse experimental approaches in live-cell imaging and protein dynamics. The protein's tolerance to circular permutation and genetic fusion has been particularly valuable for biosensor design [9]. Split-GFP systems represent another significant application, where fragments of GFP only reassemble into fluorescent complexes when fused to interacting protein partners [9]. This protein-fragment complementation assay (PCA) approach has become a powerful tool for detecting protein-protein interactions in living cells.
UnaG Experimental Protocols and Biosensor Implementation
The following detailed protocol for UnaG expression, purification, and fluorescence analysis provides researchers with a methodology for implementing this bilirubin-inducible system:
Protein Expression and Purification [5] [7]:
- Clone UnaG gene into pColdI vector with N-terminal 6xHis tag
- Transform into BL21 E. coli cells and culture in lysogeny broth with ampicillin (100 μg/mL)
- Induce expression with 0.5 mM IPTG at 16°C for 18 hours
- Harvest cells by centrifugation and lyse via sonication in potassium phosphate buffer (pH 7.5)
- Purify using HisTrap HP column with imidazole gradient (20-500 mM)
- Desalt into storage buffer using PD-10 column
Fluorescence Titration for Bilirubin Binding [5] [7]:
- Prepare UnaG samples in potassium phosphate buffer (pH 7.5)
- Titrate with bilirubin stock solution (1 mM in DMSO)
- Measure fluorescence emission at 527-532 nm with excitation at 496 nm
- Plot fluorescence intensity against bilirubin concentration
- Calculate dissociation constant (Kd) using nonlinear regression analysis
Anaerobic Imaging Applications [8]:
- Culture anaerobic bacteria (e.g., Bacteroides thetaiotaomicron) expressing UnaG
- Add bilirubin (25 μM final concentration) to growth media
- Transfer cells to anaerobic microscopy chamber
- Image using standard GFP filter sets without oxygen exposure
Molecular engineering of UnaG has created variants with altered binding affinity and fluorescence properties. For example, R112M, R132M, and double R112&132M mutations reduce polarity at bilirubin-binding sites, resulting in shifted excitation and emission profiles and modified thermal stability [7]. These engineered variants expand UnaG's utility as a quantitative biosensor for bilirubin detection, with potential clinical applications in diagnosing hyperbilirubinemia and related neurological disorders [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Experimental Reagents and Their Applications
| Reagent/Method | Function/Role | Experimental Context |
|---|---|---|
| Bilirubin (BR) | Fluorogenic ligand for UnaG [5] [8] | Prepared as 1 mM stock in DMSO [5] |
| Biliverdin (BV) | Ligand for far-red BBFPs like IFP2.0 [8] | Anaerobic multi-color imaging [8] |
| Analytical Ultracentrifugation | Determine oligomeric state [5] | Confirm monomeric state of apoUnaG and holoUnaG [5] |
| Site-Directed Mutagenesis | Engineer UnaG variants [7] | Modify bilirubin binding affinity and spectral properties [7] |
| Photon Counting Analysis | Quantify brightness states [5] | Resolve holoUnaG1 and holoUnaG2 populations [5] |
| Circular Dichroism Spectroscopy | Analyze secondary structure and stability [7] | Monitor thermal denaturation of UnaG variants [7] |
| Far-Red BBFPs (IFP2.0) | Anaerobic compatible partner for multicolor work [8] | Distinguish multiple bacterial species in co-culture [8] |
The comparative analysis of GFP and bilirubin-binding fluorescent proteins reveals a complementary relationship rather than a competitive one in the researcher's toolkit. GFP remains the superior choice for standard aerobic cellular imaging where genetic encoding and autonomous fluorescence are paramount. Its well-engineered variants and extensive validation across model systems provide a reliable platform for most fluorescence applications. Conversely, UnaG and related BBFPs offer unique capabilities for specialized investigations: anaerobic systems, rapid detection assays, and quantitative bilirubin sensing. UnaG's oxygen-independent fluorescence enables real-time imaging of previously inaccessible biological environments, particularly anaerobic microbial communities [8]. The ligand-inducible nature of UnaG provides both an advantage in temporal control and a limitation in maintaining consistent fluorescence during prolonged experiments. As protein engineering continues to advance both protein families, the strategic selection between intrinsic and induced fluorescence paradigms will depend on specific experimental requirements, environmental constraints, and measurement objectives.
The discovery and development of genetically encoded fluorescent tags have revolutionized biological imaging, enabling researchers to visualize protein localization, dynamics, and interactions in living cells and organisms [10]. While intrinsically fluorescent proteins like GFP and its variants dominate biological imaging applications, a distinct class of ligand-induced fluorescent proteins (LIFPs) has emerged with unique advantages for specialized applications [5] [10]. Among these, bilirubin-binding fluorescent proteins represent a specialized category of extrinsic fluorescent tags that derive their fluorescence from binding bilirubin, an endogenous tetrapyrrole chromophore [7] [6]. This review provides a comprehensive comparative analysis between conventional GFP-like proteins and bilirubin-binding proteins, with specific focus on UnaG, the prototypical bilirubin-inducible fluorescent protein from the Japanese eel (Anguilla japonica).
Fundamental Mechanisms: Intrinsic versus Ligand-Induced Fluorescence
GFP and Intrinsically Fluorescent Proteins
Green fluorescent protein (GFP) and its homologs are intrinsically fluorescent proteins that form their fluorophore autocatalytically from internal amino acids within an 11-stranded β-barrel structure [10]. This fluorophore formation requires molecular oxygen and can take from minutes to days depending on the specific protein, which represents a significant limitation for anaerobic applications or rapid labeling needs [10]. The fluorescence mechanism is entirely self-contained within the folded protein structure, requiring no external cofactors beyond oxygen.
Bilirubin-Binding Proteins as Ligand-Induced Fluorescent Proteins
In contrast, bilirubin-binding proteins such as UnaG belong to the fatty acid-binding protein (FABP) family and remain non-fluorescent in their apo state (apoUnaG) [5] [7]. Fluorescence is induced only upon binding of unconjugated bilirubin (UC-BR) within the protein's β-barrel structure [7] [11]. This binding occurs with high specificity for the unconjugated form of bilirubin and creates the fluorescent holoUnaG complex [11]. The fluorescence activation mechanism involves an instantaneous, oxygen-independent formation of the fluorophore, a significant advantage over GFP-family proteins [5].
Table 1: Fundamental Characteristics of Fluorescent Protein Classes
| Characteristic | GFP-like Proteins | Bilirubin-Binding Proteins |
|---|---|---|
| Fluorophore Origin | Autocatalytic from internal amino acids | External bilirubin ligand |
| Oxygen Requirement | Required for fluorophore maturation | Not required |
| Maturation Time | Several minutes to days | Instantaneous upon bilirubin binding |
| Native Structure | β-barrel | β-barrel with α-helical domains |
| Baseline Fluorescence | Constitutive | Inducible by ligand binding |
Molecular and Spectral Properties: A Detailed Comparison
Structural Characteristics
UnaG shares the fundamental β-barrel structural motif with GFP, though it belongs to the FABP family rather than the GFP superfamily [7]. The UnaG protein has a molecular weight of 15.6 kDa and comprises 139 amino acids folded into a β-barrel structure with three alpha helical domains [7]. The bilirubin binding pocket contains critical arginine residues (R112 and R132) that participate in bilirubin binding through their guanidino groups [7]. Mutation studies demonstrate that replacing these arginine residues with methionine significantly alters bilirubin binding affinity and spectral properties [7].
Spectral Properties and Quantum Efficiency
The UnaG-bilirubin complex (holoUnaG) exhibits green fluorescence with excitation and emission maxima at approximately 498 nm and 527 nm, respectively [7] [11]. The quantum efficiency of holoUnaG is remarkably high at approximately 50%, comparable to enhanced GFP (EGFP), one of the brightest GFP mutants [5]. Recent research has revealed that holoUnaG exists in two distinct fluorescence states (holoUnaG1 and holoUnaG2) that reach equilibrium after initial complex formation, with a molecular brightness ratio of 1:3.9 and equilibrium population ratio of 6:4 [5].
Table 2: Spectral Properties Comparison
| Parameter | EGFP | UnaG | GymFP |
|---|---|---|---|
| Excitation Maximum (nm) | 488 | 498 | 496 |
| Emission Maximum (nm) | 507 | 527 | 532 |
| Quantum Yield | 0.60 | ~0.50 | Not reported |
| Extinction Coefficient (mM⁻¹cm⁻¹) | 56 | Not reported | Not reported |
| Brightness (Quantum Yield × Extinction Coefficient) | 33.6 | Comparable to EGFP | Not reported |
| Maturation Time | Hours | Instantaneous | Instantaneous |
Experimental Applications and Protocols
Bilirubin Detection and Quantification
The high specificity of UnaG for unconjugated bilirubin has been harnessed for clinical detection applications. A robust protocol for serum bilirubin measurement involves incubating serum samples with purified UnaG protein and measuring fluorescence intensity, which shows linear correlation with bilirubin concentrations from 0 to 70 mg/dL [11]. This method demonstrates exceptional precision with coefficients of variation <10% and remains unaffected by common serum interferents like hemoglobin or lipid emulsion [11]. The technique has been successfully validated in newborn serum samples, demonstrating strong correlation (r = 0.943, P < 0.001) with conventional bilirubin oxidase methods [11].
Protein Engineering and Mutant Development
Protein engineering efforts have generated UnaG variants with modified properties. The R112M, R132M, and R112&132M mutants show altered spectral properties, with the double mutant exhibiting a pronounced blue shift in excitation (504 nm) and emission (542 nm) peaks [7]. These mutations also affect thermal stability, with the R112M mutant demonstrating the lowest melting temperature (53.8°C) among the variants [7]. Another engineered variant, eUnaG, exhibits approximately twice the fluorescence intensity of wild-type UnaG through modification of the valine amino acid at position 2 [7].
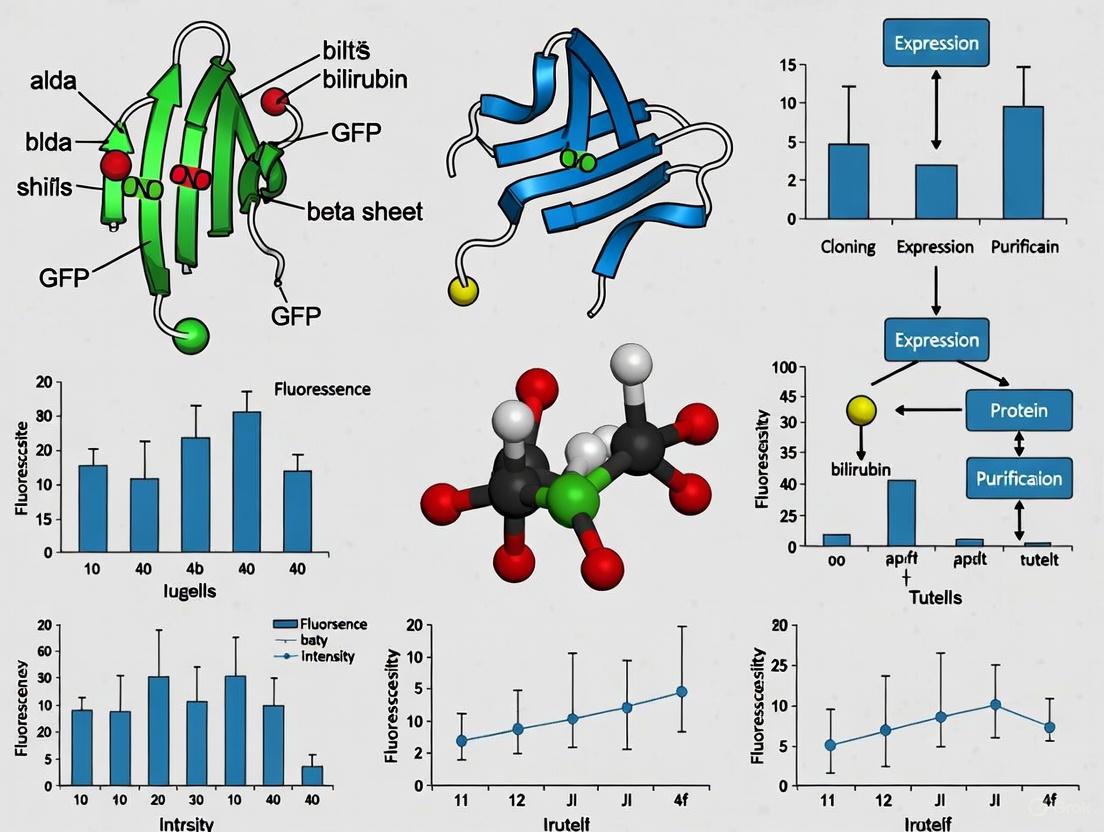
Diagram 1: UnaG Fluorescence Activation Pathway. This diagram illustrates the transition from non-fluorescent ApoUnaG to the two distinct fluorescent states of HoloUnaG upon bilirubin binding, culminating in an equilibrium state between the two forms.
Advantages and Limitations in Research Applications
Unique Advantages of Bilirubin-Binding Proteins
- Oxygen-Independent Maturation: Unlike GFP, UnaG fluorescence activation doesn't require molecular oxygen, enabling applications in anaerobic conditions and hypoxic environments [5].
- Instantaneous Activation: Fluorescence develops immediately upon bilirubin binding, unlike GFP variants that require time for fluorophore maturation [5].
- Endogenous Fluorophore Utilization: Can leverage naturally occurring bilirubin in biological systems, potentially enabling monitoring of heme catabolism pathways [12].
- Specific Biosensing Capability: The specific binding to unconjugated bilirubin enables direct development as a biosensor for this clinically relevant molecule [11].
Practical Limitations and Challenges
- Dependence on Bilirubin Availability: Fluorescence is contingent upon bilirubin presence and proper delivery to the cellular compartment of interest [7].
- Potential Endogenous Interference: In systems with natural bilirubin fluctuations, background signals may complicate interpretation [12].
- Limited Color Palette: Currently, bilirubin-binding proteins are primarily green-emitting, unlike the broad color palette of GFP variants [10].
- Relatively New Technology: Less established and characterized compared to the extensive toolkit of GFP variants and applications [6].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for Bilirubin-Binding Protein Work
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Expression Vectors | pColdI-UnaG, pTolT-UnaG, pGEX-UnaG | Recombinant protein expression in E. coli systems [5] [7] |
| Chromophore | Unconjugated bilirubin (UC-BR) | Fluorophore ligand for activating fluorescence [5] [7] |
| Purification Systems | HisTrap HP column (for 6×His-tagged UnaG) | Affinity purification of recombinant proteins [5] |
| Mutagenesis Kits | Phusion Site-Directed Mutagenesis Kit | Creating specific UnaG mutants [7] |
| Spectroscopic Tools | Fluorescence spectrophotometer (e.g., Hitachi F-7100) | Measuring excitation/emission spectra and binding kinetics [7] |
| Structural Analysis | Circular Dichroism (CD) spectroscopy | Assessing secondary structure and thermal stability [7] |
Diagram 2: UnaG Protein Engineering Workflow. This experimental workflow outlines the key steps in generating and characterizing UnaG variants, from gene synthesis to spectroscopic analysis.
Bilirubin-binding proteins represent a distinct class of extrinsic fluorescent tags with unique mechanisms and applications that complement conventional GFP-like proteins. Their oxygen-independent, instantaneous activation and specific binding to an endogenous ligand provide unique advantages for specialized applications including hypoxia imaging, bilirubin sensing, and rapid protein tagging [5] [11]. While the GFP toolkit offers broader color options and more established protocols, bilirubin-binding proteins like UnaG and newly discovered homologs such as GymFP from the moray eel (Gymnothorax zonipectis) expand the fluorescent protein arsenal with novel functionalities [6]. Future directions include engineering brighter variants, expanding the spectral range, and developing optimized delivery systems for bilirubin in cellular environments. For researchers considering these tools, bilirubin-binding proteins offer the greatest advantage in applications involving hypoxic conditions, rapid labeling requirements, or when leveraging bilirubin as a biological signal.
Fluorescent proteins (FPs) are indispensable tools in biological research, enabling the visualization of cellular processes in real time. Despite their similar functional output—the emission of light—these proteins achieve fluorescence through fundamentally different biochemical pathways. This guide provides a structured comparison between two principal classes of fluorescent proteins: those with autocatalytic maturation, exemplified by the Green Fluorescent Protein (GFP), and those with cofactor-dependent activation, such as bilirubin-binding proteins including UnaG. The core distinction lies in the origin of their chromophores; GFP self-assembles its chromophore from its own amino acids, while UnaG must bind an externally synthesized ligand, bilirubin, to become fluorescent. This article objectively compares their performance, detailing underlying mechanisms, experimental workflows, and key reagent solutions for researchers and drug development professionals.
Fundamental Mechanisms of Chromophore Formation
The genesis of the light-emitting chromophore is the critical event that confers fluorescence. The pathways for GFP-like proteins and bilirubin-binding FPs are distinct, involving either an internal synthesis or an external binding process.
Autocatalytic Maturation in Green Fluorescent Protein (GFP) The GFP chromophore is formed post-translationally from three consecutive amino acids within its own polypeptide chain (Ser65–Tyr66–Gly67 in Aequorea victoria GFP) [13]. This process is autocatalytic, requiring only molecular oxygen and proceeding without the need for external enzymes or cofactors [13]. The generally accepted mechanism involves a series of internal reactions within the folded protein: cyclization, dehydration, and oxidation [14]. Theoretical studies using hybrid density functional theory (B3LYP) indicate that the rate-limiting cyclization step is initiated by the deprotonation of the Gly67 amide nitrogen, a process facilitated by the protein's internal environment, including residues Arg96 and Glu222 acting as a general base [14]. This creates an extended π-conjugation system within the imidazolinone ring, enabling light absorption and emission.
Cofactor-Dependent Activation in Bilirubin-Binding Proteins (UnaG) In stark contrast, fluorescent proteins like UnaG, discovered in the Japanese eel Anguilla japonica, belong to the fatty acid binding protein family and are not intrinsically fluorescent [7] [6]. Their fluorescence is induced upon the highly specific, non-covalent binding of an external chromophore, bilirubin [7]. Bilirubin is a linear tetrapyrrole molecule produced from the catabolism of heme in vertebrates [7]. The apo form of UnaG (apoUnaG) does not fluoresce. Upon binding bilirubin, it forms the holo complex (holoUnaG), which emits bright green light (peak emission at 527 nm) [7]. The binding affinity is high, with dissociation constants ((K_d)) in the nanomolar range, and is stabilized by specific amino acid interactions, such as arginine residues R112 and R132 in wild-type UnaG [7].
The diagrams below summarize these two fundamental pathways and their key characteristics.
Figure 1: Comparative chromophore genesis pathways. GFP undergoes an internal, multi-step maturation process, while UnaG requires the binding of an externally produced bilirubin molecule to become fluorescent.
Experimental Analysis & Workflows
Studying these proteins requires tailored experimental approaches to probe their distinct maturation, stability, and spectral properties.
Probing GFP's Autocatalytic Mechanism
The study of GFP maturation often involves site-directed mutagenesis to alter key chromophore residues, followed by spectroscopic analysis and theoretical calculations.
- In Silico Mechanism Studies: Cluster models based on X-ray crystal structures (e.g., PDB: 2AWJ) are used with hybrid density functional theory (e.g., B3LYP) to calculate potential energy profiles for cyclization. This allows researchers to compare the energy barriers of different proposed mechanisms, such as cyclization initiated by deprotonation of the Gly67 amide nitrogen versus the Tyr66 α-carbon [14].
- In-Gel Fluorescence Detection: A key practical protocol involves detecting GFP fluorescence directly in SDS-PAGE gels, known as In-Gel Fluorescence (IGF). The robust β-barrel structure of GFP and its derivatives allows them to retain fluorescence even after denaturation in SDS sample buffer at high temperatures [15].
- Workflow: Cell extracts expressing GFP-fusion proteins are prepared with standard Laemmli buffer, heated (95°C, 5-10 min), and loaded onto a polyacrylamide gel. After electrophoresis, the gel is immediately imaged using a standard fluorescence imager with appropriate filters (e.g., 488 nm excitation/510 nm emission) without the need for protein transfer or antibody staining [15].
- Application: This protocol is used for rapid verification of fusion protein integrity, quantification, and screening, offering high sensitivity and a broad dynamic range with minimal background [15].
Characterizing UnaG-Bilirubin Interaction
Analyzing bilirubin-binding FPs focuses on the protein-ligand interaction, structural changes upon binding, and tuning of biophysical properties.
- Ligand Binding and Spectroscopic Characterization: The core protocol involves titrating a purified, non-fluorescent apoUnaG protein sample with increasing concentrations of bilirubin and monitoring the emergence of fluorescence.
- Workflow: Purified apoUnaG (from recombinant expression in E. coli) is diluted in a suitable buffer. A concentrated stock solution of bilirubin (in DMSO or NaOH) is added in increments. After each addition, the fluorescence emission spectrum (e.g., 500-600 nm, with excitation at ~490 nm) is recorded. The fluorescence intensity at the emission maximum (~530 nm) is plotted against the bilirubin concentration to determine the dissociation constant ((K_d)) [7].
- Mutagenesis to Modulate Function: Site-directed mutagenesis of key bilirubin-binding residues is performed to engineer UnaG variants. For instance, mutating arginine residues (R112, R132) to methionine reduces polarity in the binding pocket, which can alter the excitation/emission peaks, binding affinity ((K_d)), and thermal stability of the protein [7].
- Structural Stability Assessment: The impact of bilirubin binding on protein structure is assessed using Circular Dichroism (CD) Spectroscopy in the far-UV region (190-250 nm). This technique compares the secondary structure of apoUnaG (often showing low ellipticity and poor defined structure) with holoUnaG (showing characteristic α-helical and β-sheet signatures), confirming that ligand binding induces proper folding [7].
The following diagram illustrates a generalized experimental workflow for characterizing a bilirubin-inducible fluorescent protein like UnaG.
Figure 2: Core workflow for characterizing a bilirubin-inducible fluorescent protein. The process from gene identification to biophysical analysis enables the determination of key functional parameters.
Comparative Performance Data
The table below summarizes the key characteristics and performance metrics of GFP and UnaG, highlighting their differences for research applications.
Table 1: Performance comparison of autocatalytic GFP and cofactor-dependent UnaG.
| Feature | Green Fluorescent Protein (GFP) | Bilirubin-Binding Protein (UnaG) |
|---|---|---|
| Chromophore Origin | Autocatalytic (Internal Ser-Tyr-Gly) [13] | Exogenous ligand (Bilirubin) [7] |
| Maturation Requirement | Molecular oxygen (O₂) [13] | Binding of bilirubin [7] |
| Typical Excitation/Emission | Ex ~488 nm / Em ~509 nm (for EGFP) [13] | Ex ~496 nm / Em ~527 nm [6] |
| Fluorescence Quantum Yield | 0.60 (EGFP) [13] | 0.51 (Wild-type UnaG, varies by mutant) [7] |
| Extinction Coefficient (ε) | ~55,000 M⁻¹cm⁻¹ (EGFP) [13] | Information not specified in results |
| Ligand Dissociation Constant ((K_d)) | Not applicable | Nanomolar range (e.g., ~98 nM for R132M UnaG mutant) [7] |
| Key Advantage | No external cofactor needed; self-sufficient | High specificity for bilirubin; usable as biosensor |
| Key Limitation | Slow maturation at lower temperatures; sensitivity to pH (wtGFP) | Dependent on bilirubin availability and concentration |
The Researcher's Toolkit: Essential Reagents & Materials
Successful experimentation with these fluorescent proteins requires a specific set of reagents and materials. The following table details key solutions for the featured experiments.
Table 2: Key research reagent solutions for studying fluorescent protein maturation and function.
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Site-Directed Mutagenesis Kit | Introduces point mutations into gene sequences to study chromophore formation or ligand binding. | Engineering GFP chromophore mutants (S65T) [13] or UnaG binding pocket mutants (R112M) [7]. |
| pBAD/His Expression Vector | Allows tightly controlled, inducible recombinant expression of proteins in E. coli. | Expression of UnaG and its mutant variants for purification [7]. |
| Bilirubin Stock Solution | The essential ligand for activating fluorescence in UnaG and related proteins. | Titration into apoUnaG for fluorescence characterization and (K_d) determination [7]. |
| Fluorescence Spectrophotometer | Measures the excitation and emission spectra of fluorescent samples; quantifies intensity. | Determining the (λ{ex}/λ{em}) maxima and quantum yield of GFP and UnaG [7]. |
| Circular Dichroism (CD) Spectrometer | Analyzes the secondary structure and stability of proteins in solution. | Confirming the structural change in UnaG upon bilirubin binding and assessing thermal stability [7]. |
| SDS-PAGE Gel Electrophoresis System | Separates proteins by molecular weight; used with in-gel fluorescence (IGF) detection. | Rapidly verifying the expression and integrity of GFP-fusion proteins directly in gels [15]. |
Discussion & Research Implications
The choice between autocatalytic and cofactor-dependent fluorescent proteins is fundamental and depends on the research context. Each system presents unique advantages and challenges that direct their application.
GFP and its engineered variants (e.g., EGFP, sfGFP) offer a self-contained system. Their ability to fold and form a chromophore autonomously in diverse cellular environments makes them exceptionally versatile for general-purpose tagging and reporting in live cells [13]. However, their maturation can be slow and is sensitive to the cellular environment, such as pH and redox potential [13]. The robust, covalent nature of its chromophore also allows for direct detection in denaturing SDS-PAGE gels (IGF), a significant advantage for biochemical analyses [15].
In contrast, the bilirubin-dependent fluorescence of UnaG defines a specialized niche. Its absolute requirement for an endogenous metabolite makes it a powerful biosensor for bilirubin, with direct applications in diagnosing liver function and jaundice [7]. The ligand-induced fluorescence provides a "switch-on" mechanism, eliminating background from unbound apo-protein. This property is being exploited to develop rapid, inexpensive, and specific clinical detection methods for bilirubin [7]. Furthermore, the discovery of UnaG homologs in other eels, like GymFP from the moray eel, demonstrates the natural diversity and evolutionary expansion of this protein family [6].
From a protein engineering perspective, the two systems offer different handles for optimization. GFP is typically engineered by mutating residues around the chromophore to alter its color, brightness, and stability [13]. UnaG, however, can be tuned by mutating residues involved in bilirubin binding (e.g., R112, R132) to modulate its affinity, spectral shift, and thermal stability, as demonstrated in recent studies [7].
In conclusion, the dichotomy between autocatalytic and cofactor-dependent maturation underpins the functional diversity of the fluorescent protein toolkit. While GFP remains a cornerstone for generic cellular imaging, bilirubin-binding proteins like UnaG are emerging as highly specific biosensors, opening new avenues in biomedical diagnostics and fundamental research.
Fluorescent proteins (FPs) have revolutionized molecular and cellular biology, enabling researchers to visualize dynamic processes within living systems. Since the discovery of the original Green Fluorescent Protein (GFP) from Aequorea victoria, the FP toolbox has expanded dramatically to include proteins with diverse spectral properties and biochemical characteristics. These proteins can be broadly categorized by their chromophore type and source. The classic GFP-like proteins form their chromophores through an autocatalytic process requiring oxygen, while a newer class of proteins binds to exogenous or endogenous fluorogenic chromophores such as bilirubin, biliverdin, or flavins. This diversity provides researchers with a rich palette of tools for imaging, each with unique advantages and limitations in terms of brightness, spectral range, and environmental requirements.
The quest for spectral diversity is driven by the need for multi-color imaging, deeper tissue penetration, and compatibility with different microscope systems and experimental conditions. Brightness, a product of a protein's extinction coefficient and quantum yield, is a critical parameter determining the signal-to-noise ratio in fluorescence imaging. This guide provides an objective comparison of the performance characteristics across different fluorescent protein systems, with a specific focus on GFP versus bilirubin-binding fluorescent proteins, to assist researchers in selecting the optimal probes for their experimental needs.
Performance Comparison of Fluorescent Protein Systems
The table below summarizes the key photophysical properties of major fluorescent protein classes, providing a quantitative basis for comparison.
Table 1: Photophysical Properties of Major Fluorescent Protein Classes
| Protein Class | Example Protein | Excitation Max (nm) | Emission Max (nm) | Extinction Coefficient (M⁻¹cm⁻¹) | Quantum Yield | Molecular Brightness* | Oxygen Requirement |
|---|---|---|---|---|---|---|---|
| GFP-like | sfGFP | 485 | 510 | 83,000 [16] | 0.65 [16] | 53,950 | Yes |
| GFP-like (Enhanced) | YuzuFP | 485 | 510 | ~90,000 [16] | ~0.72 [16] | ~64,800 | Yes |
| Bilirubin-binding | UnaG | 498 | 527 | 77,900 [5] | 0.51 [5] | 39,729 | No [8] [17] |
| Bilirubin-binding | GymFP | 496 | 532 | Not specified | Not specified | Not specified | No [6] |
| Biliverdin-binding | IFP2.0 | 690 | 711 | 82,000 [8] | 0.08 [8] | 6,560 | No [8] |
| Biliverdin-binding (Triad) | BDFP1.1:3.1:1.1 | ~650-700 (FR) | 722 | Not specified | Not specified | High (in cells) [18] | No [18] |
| Flavin-binding | miniGFP1/2 | 450 | 499 | ~46,000 [19] | ~0.60 [19] | ~27,600 | No [19] |
Note: Molecular Brightness is calculated as the product of Extinction Coefficient and Quantum Yield, normalized to the value for EGFP (often set to 1.0 in literature). The values here are approximate for cross-comparison. FR = Far-Red light.
Analysis of Performance Data
The quantitative data reveals clear trade-offs between different fluorescent protein systems. GFP-like proteins, particularly enhanced variants like YuzuFP, generally offer the highest molecular brightness in the green-yellow spectrum, with improvements achieved through mutations that optimize chromophore interaction networks [16]. For instance, the H148S mutation in YuzuFP increases persistent hydrogen bonding with the chromophore and water residency time, resulting in a 1.5-fold brightness increase and 3-fold enhanced photobleaching resistance compared to sfGFP [16].
Bilirubin-binding proteins like UnaG and GymFP provide strong green fluorescence with the significant advantage of oxygen-independent maturation [6] [8] [17]. While UnaG's quantum yield (0.51) is moderately high, its practical brightness in mammalian cells is comparable to GFP because it utilizes endogenous bilirubin [5] [17]. These proteins exhibit instantaneous fluorescence upon bilirubin binding without the delay required for GFP chromophore oxidation [5].
Near-infrared systems based on biliverdin binding, such as the BDFP triad, offer emission in the tissue-penetrating spectral window (700-770 nm) but often at the cost of reduced quantum yield, as seen with IFP2.0 (0.08) [18] [8]. Engineered systems that employ FRET between biliverdin- and phytochromobilin-binding domains can overcome this limitation, achieving both large red-shift and high brightness in mammalian cells [18].
Experimental Protocols for Key Applications
Protocol 1: Live-Cell Anaerobic Imaging Using Bilirubin-Binding Proteins
Purpose: To enable fluorescence imaging of protein dynamics and localization in obligate anaerobic bacteria, such as those found in the gut microbiome, where conventional GFP-like proteins fail to mature [8].
Materials:
- Bacterial Strains: Bacteroides thetaiotaomicron (B. theta) or other anaerobic species.
- Plasmids: Expression vectors encoding UnaG (cytoplasmic or fusion constructs).
- Ligand Solution: 25 mM bilirubin stock in DMSO [8].
- Growth Media: Rich media (tryptone-yeast extract-glucose) and minimal media with carbohydrate source (0.05% w/v glucose or maltose) [8].
- Anaerobic Chamber: Coy chamber or equivalent maintaining anaerobic atmosphere (typically 85% N₂, 10% CO₂, 5% H₂) [8].
Method:
- Transformation: Introduce the UnaG expression plasmid into B. theta via electroporation or conjugation.
- Culture Inoculation: Pick single colonies and inoculate 5 mL of rich media containing appropriate antibiotic. Grow anaerobically at 37°C for 24-48 hours.
- Back Dilution: Dilute the culture 1:100 into fresh minimal media containing 25 µM bilirubin [8].
- Anaerobic Growth: Grow cells anaerobically to mid-log phase (OD₆₀₀ ~0.4-0.6).
- Microscopy Preparation: Transfer a small aliquot of cells (~2 µL) to a glass coverslip. Seal with a second coverslip using epoxy adhesive to maintain anaerobic conditions during imaging [8].
- Image Acquisition: Image cells using standard GFP filter sets (excitation ~470-490 nm, emission ~500-540 nm) on an epifluorescence or confocal microscope.
Technical Notes: Bilirubin is light-sensitive and should be handled in dim light. DMSO concentrations in media should be kept below 1% to avoid toxicity. For two-color anaerobic imaging, IFP2.0 (a biliverdin-binding protein) can be co-expressed and visualized using far-red filter sets [8].
Protocol 2: Characterizing Bilirubin Binding Kinetics and Fluorescence States
Purpose: To quantitatively analyze the binding interaction between UnaG and its bilirubin ligand, including the discovery and characterization of distinct fluorescent states [5].
Materials:
- Purified Protein: Recombinant 6xHis-tagged UnaG protein purified via immobilized metal affinity chromatography (IMAC) [5].
- Ligand: 1 mM bilirubin stock solution in DMSO (store at -20°C protected from light) [5].
- Buffers: 20 mM potassium phosphate buffer, pH 7.5 [5].
- Instrumentation: Spectrofluorometer, analytical ultracentrifuge [5].
Method:
- Protein Preparation: Express and purify UnaG from E. coli. Determine protein concentration spectrophotometrically.
- Fluorescence Time Course: Rapidly mix UnaG (final concentration 1 µM) with bilirubin (final concentration 1.5 µM) in potassium phosphate buffer. Immediately monitor fluorescence intensity (excitation 500 nm, emission 527 nm) over time [5].
- State Differentiation: Observe the biphasic fluorescence increase, indicative of the formation of two distinct states: the initial holoUnaG1 and the brighter, final holoUnaG2 [5].
- Equilibrium Analysis: After signal stabilization, measure the excitation and emission spectra of the mature holoUnaG complex.
- Quenching Experiments: Add a known concentration of a competing bilirubin-binding molecule (e.g., human serum albumin) to the mature holoUnaG complex and monitor fluorescence decrease to estimate dissociation rates [5].
- Oligomeric State Verification: Perform analytical ultracentrifugation sedimentation equilibrium analysis (AUC-SE) to confirm UnaG exists as a monomer in solution [5].
Technical Notes: The molecular brightness ratio of holoUnaG1 to holoUnaG2 is approximately 1:3.9, with an equilibrium population ratio of about 6:4 [5]. The intra-molecular reaction converting the two states is reversible and associated with a change in the chromophore environment rather than a chemical modification of bilirubin itself [5].
Signaling Pathways and Experimental Workflows
The following diagram illustrates the fundamental mechanistic differences in how GFP-like proteins and bilirubin-binding proteins achieve fluorescence, which underpins their distinct experimental applications.
Diagram 1: Contrasting fluorescence activation pathways between GFP-like and bilirubin-binding proteins. The key distinction is the oxygen-dependent chromophore maturation in GFP versus the instantaneous, oxygen-independent ligand binding in bilirubin-binding proteins.
Research Reagent Solutions
This table outlines essential materials and their applications for working with different fluorescent protein systems, serving as a quick-reference guide for experimental planning.
Table 2: Essential Research Reagents for Fluorescent Protein Applications
| Reagent / Material | Function / Application | Example Use Cases |
|---|---|---|
| Bilirubin | Fluorogenic ligand for UnaG, GymFP, and related bilirubin-binding FPs [5] [8] | Anaerobic live-cell imaging [8]; BR distribution studies [5] |
| Biliverdin | Fluorogenic ligand for near-infrared FPs (IFP2.0, BDFPs) [18] [8] | Deep-tissue imaging; multi-color anaerobic studies with UnaG [8] |
| Flavin Mononucleotide (FMN) | Endogenous chromophore for flavin-binding FPs (FbFPs, miniGFPs) [19] [17] | Hypoxia and anaerobic imaging; metal ion biosensing [19] |
| Anaerobic Chamber | Maintains oxygen-free environment for culture and imaging of obligate anaerobes [8] | Gut microbiome research; studies of anaerobic pathogens [8] |
| Superfolder GFP (sfGFP) | Robust, fast-folding GFP variant; baseline for engineering and comparison [16] | General protein tagging in aerobic conditions; reference standard for brightness [16] |
| HaloTag System | Chemical-genetic tagging system for anaerobic single-particle tracking [8] | Protein dynamics in anaerobic bacteria when expressed FPs are unsuitable [8] |
The expanding spectral diversity of fluorescent proteins provides researchers with an increasingly sophisticated toolkit for biological imaging. GFP-like proteins continue to be optimized for brightness and photostability through rational design and molecular dynamics-guided engineering, as demonstrated by variants like YuzuFP [16]. Simultaneously, bilirubin- and biliverdin-binding proteins offer unique capabilities for anaerobic imaging and deeper tissue penetration, albeit sometimes with compromises in absolute quantum yield.
The choice between these systems is fundamentally application-dependent. For standard aerobic cell culture with maximal brightness in the green spectrum, enhanced GFP variants remain superior. However, for investigating anaerobic microbiomes, hypoxic tumor environments, or for deep-tissue imaging requiring near-infrared emission, bilin-binding proteins are indispensable [8] [17]. Future directions will likely focus on engineering brighter near-infrared variants, developing more sensitive biosensors based on environmental sensitivity of these proteins, and creating new systems with tailored photophysical properties for advanced microscopy techniques such as super-resolution imaging and cryo-electron microscopy.
From Lab to Clinic: Practical Applications and Innovative Uses
Live-cell imaging represents a cornerstone of modern biological research, enabling scientists to visualize the intricate dynamics of proteins and cellular processes in real-time. For decades, the green fluorescent protein (GFP) and its spectral variants have served as the workhorse tools in this field, revolutionizing our ability to track protein localization, interaction, and function in living systems. However, GFP possesses inherent limitations that restrict its application across all biological contexts. This guide provides a comprehensive objective comparison between conventional GFP-based systems and emerging bilirubin-binding fluorescent proteins (BBFPs), equipping researchers with the experimental data and methodologies necessary to select the optimal tool for their live-cell imaging investigations, particularly under challenging physiological conditions.
Technical Performance Comparison: GFP vs. BBFPs
Table 1: Quantitative Performance Comparison of Fluorescent Proteins for Live-Cell Imaging
| Feature/Parameter | GFP (mEGFP) | UnaG (BBFP) | IFP2.0 (BBFP) |
|---|---|---|---|
| Chromophore Formation | Oxygen-dependent maturation [8] [20] | Oxygen-independent bilirubin binding [8] [21] | Oxygen-independent biliverdin binding [8] [21] |
| Excitation/Emission Max (nm) | ~488/~510 [22] | ~495/~510 [8] [23] | Far-red excitation/emission [8] |
| Relative Brightness | Bright (benchmark) | Similar to GFP [8] | Dimmer than GFP or UnaG [8] |
| Ligand Requirement | None | Bilirubin (BR) [8] | Biliverdin (BV) [8] |
| Ligand Permeability | Not Applicable | Cell-permeable [8] | Cell-permeable [8] |
| Anaerobic Function | No [8] [20] | Yes [8] [21] [23] | Yes [8] [21] |
| pKa (Acid Sensitivity) | ~6.0 [20] | Data not fully established | Data not fully established |
| Multicolor Imaging | Excellent with variants (e.g., CFP, YFP) [22] | Yes (Green); Orange ligand available [23] | Yes (Far-red) [8] |
Experimental Protocols for Anaerobic Live-Cell Imaging
The following detailed methodology outlines the implementation of BBFPs for live-cell imaging of anaerobic bacteria, as substantiated by key research.
Protocol: Implementing BBFPs in Gut Commensal Bacteria
1. Bacterial Strain Generation and Culture Conditions
- Strains and Plasmids: Utilize Bacteroides thetaiotaomicron (B. theta) or other genetically tractable anaerobes. Clone UnaG or IFP2.0 coding sequences into appropriate expression vectors [8].
- Anaerobic Growth: Culture bacteria in rich media (e.g., tryptone-yeast extract-glucose) within an anaerobic chamber (e.g., Coy chamber) at 37°C. For experiments, back-dilute cultures into defined minimal media with a relevant carbohydrate source (e.g., 0.05% w/v glucose) [8].
- Ligand Preparation: Prepare stock solutions of bilirubin (br) or biliverdin (bv) in DMSO. Supplement growth media with final concentrations of 25 µM bilirubin for UnaG or 2.5 µM biliverdin for IFP2.0 [8].
2. Live-Cell Anaerobic Imaging
- Sample Preparation: Grow cells to early- to mid-log phase. For imaging, seal cells between coverslips using epoxy resin inside the anaerobic chamber to maintain an oxygen-free environment throughout the imaging process [8].
- Microscopy: Perform imaging at room temperature using standard epifluorescence or confocal microscopes. For UnaG, use GFP filter sets (Ex ~485 nm). For IFP2.0, use far-red filter sets [8].
- Controls: Always include wild-type cells not expressing the BBFP but incubated with the same ligand concentration to account for any background or autofluorescence [8] [23].
Protocol: Validation and Functional Assays
1. Ligand Binding and Specificity
- In Vitro Titration: Purify the BBFP and perform fluorescence titration with increasing concentrations of its ligand (br or bv) to determine the dissociation constant (Kd). For the novel orange UnaG ligand, a Kd of 3 nM was measured [23].
- Competitive Assay: To confirm binding occurs in the native bilirubin pocket, perform a competition assay by adding excess native ligand (e.g., bilirubin) and observe the reduction in fluorescence from the new ligand pair [23].
2. Toxicity and Growth Curves
- Monitor bacterial growth via absorbance at OD600 in the presence and absence of ligands to ensure no adverse effects on cell viability or division. Research has shown that ligands like the benzothiazole-based compound "2" at 2.5 µM do not affect the growth kinetics of B. theta [23].
Visualization of Signaling Pathways and Workflows
Diagram 1: Chromophore Maturation Mechanisms
Diagram 2: Experimental Workflow for Anaerobic Imaging with BBFPs
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Live-Cell Imaging with GFP and BBFPs
| Reagent / Material | Function / Application | Example Use-Case |
|---|---|---|
| Anaerobic Chamber | Creates and maintains oxygen-free environment for culture and imaging. | Essential for culturing obligate anaerobes like Bacteroides spp. and for all anaerobic imaging steps [8]. |
| Bilirubin (BR) | Fluorogenic ligand for UnaG BBFP. | Added to media (e.g., 25 µM) to activate green fluorescence in UnaG-expressing cells [8]. |
| Biliverdin (BV) | Fluorogenic ligand for IFP2.0 and other far-red BBFPs. | Added to media (e.g., 2.5 µM) to activate far-red fluorescence [8]. |
| Benzothiazole Ligand "2" | Synthetic orange-emitting ligand for UnaG. | Used at ~2.5 µM for labeling cells in the 540/570 nm channel for multicolor experiments [23]. |
| Epoxy Sealant | Chemically inert sealing agent for microscopy slides. | Seals samples under anaerobic conditions prior to imaging to prevent oxygen ingress [8]. |
| Custom Donor Vectors | Plasmid constructs for CRISPR/Cas9 knock-in of fluorescent tags. | For endogenous tagging of low-abundance proteins in human cells (e.g., PER2, CRY1) [24]. |
The choice between GFP and bilirubin-binding fluorescent proteins is not a matter of simple superiority but of application-specific suitability. GFP and its variants remain powerful, robust, and largely unparalleled for standard live-cell imaging in aerobic systems, offering a bright, well-characterized, and convenient platform.
However, for investigations venturing into anaerobic environments—such as the gut microbiome, soil ecosystems, or other hypoxic tissues—BBFPs provide a critical and enabling technology. Their oxygen-independent maturation, cell-permeable fluorogenic ligands, and growing color palette make them the definitive choice for these challenging yet biologically crucial contexts. As research continues to illuminate the complex dynamics of life in all its environments, the availability of diverse and specialized tools like BBFPs will be paramount to driving scientific discovery forward.
The development of genetically encoded biosensors represents one of the most significant advancements in molecular biology, enabling real-time monitoring of cellular processes with high spatial and temporal resolution. While traditional Green Fluorescent Protein (GFP) and its variants have revolutionized cell biology, a new class of bilirubin-inducible fluorescent proteins (FPs) derived from eels offers distinctive advantages for specific biosensing applications [25]. These unique FPs, belonging to the fatty acid binding protein (FABP) family, utilize endogenous bilirubin as a fluorogen and exhibit oxygen-independent maturation, making them particularly valuable for imaging under hypoxic conditions and for detecting specific metabolites [7] [25]. This review provides a comprehensive comparison between GFP-based and bilirubin-binding FP-based biosensors, examining their molecular mechanisms, performance characteristics, and practical applications in biomedical research and drug development.
Molecular Mechanisms and Fundamental Properties
GFP and Its Variants: Structure-Driven Fluorescence
Green Fluorescent Protein and its engineered variants function through an autocatalytically formed chromophore within a protective β-barrel structure [26]. The GFP chromophore derives from a tripeptide sequence (Ser-Tyr-Gly) that undergoes cyclization, dehydration, and oxidation to form the 4-(p-hydroxybenzylidene)-5-imidazolidinone fluorophore [26]. This maturation process requires molecular oxygen, which can limit applications in anaerobic or hypoxic environments [25]. The extensive engineering of GFP has produced variants spanning the visible spectrum, including blue (BFP), cyan (CFP), and yellow (YFP) fluorescent proteins, enabling multi-color imaging and FRET-based sensing approaches [26].
Bilirubin-Binding FPs: Ligand-Induced Fluorescence
In contrast, bilirubin-binding FPs such as UnaG (from Anguilla japonica) and the recently discovered GymFP (from Gymnothorax zonipectis) belong to the FABP family and remain non-fluorescent in their apo state [6] [7]. Their fluorescence is induced by bilirubin binding—a linear tetrapyrrole produced during heme catabolism [7] [5]. The unique Gly-Pro-Pro (GPP) motif conserved in these fluorescent FABPs serves as a structural element that protects bilirubin from solvent quenching, enabling bright fluorescence [27]. Unlike GFP, the fluorophore formation is instantaneous and oxygen-independent, as the chromophore is pre-formed rather than synthesized de novo [25].
Table 1: Fundamental Properties of Fluorescent Protein Classes
| Property | GFP-like Proteins | Bilirubin-Binding FPs |
|---|---|---|
| Chromophore Origin | Autocatalytic from protein sequence | Exogenous bilirubin ligand |
| Maturation Requirement | Oxygen-dependent | Oxygen-independent |
| Maturation Time | Hours | Instantaneous upon bilirubin binding |
| Native Structure | β-can fold | β-barrel (FABP family) |
| Molecular Weight | 25-27 kDa | ~15-16 kDa |
| Key Structural Motif | Ser-Tyr-Gly tripeptide | Gly-Pro-Pro motif |
Diagram 1: Fundamental differences in biosensor mechanisms between GFP-based and bilirubin-binding fluorescent proteins.
Performance Comparison and Experimental Data
Spectral Properties and Brightness Characteristics
Direct comparison of spectroscopic properties reveals distinct advantages for each FP class. UnaG exhibits excitation and emission maxima at approximately 498 nm and 527 nm, respectively, with a quantum efficiency of nearly 50% when bound to bilirubin—comparable to enhanced GFP but with the advantage of instantaneous activation [7] [5]. Recent studies have identified that the holoUnaG complex exists in two distinct fluorescence states (holoUnaG1 and holoUnaG2) with a molecular brightness ratio of 1:3.9, suggesting potential for engineering brighter variants [5]. GymFP from moray eel shows similar spectral characteristics with excitation at 496 nm and emission at 532 nm [6].
The small size of bilirubin-binding FPs (~15 kDa versus 25-27 kDa for GFP-like FPs) provides significant advantages for genetic fusion constructs, reducing the potential for steric interference with target protein function [25]. This compact size is particularly beneficial for viral vector-based gene delivery where genetic payload capacity is limited [19].
Environmental Stability and Sensing Capabilities
Bilirubin-binding FPs demonstrate remarkable stability across various environmental conditions. Engineering studies have shown that UnaG mutants maintain function under physiological detection conditions, with certain point mutations (R112M, R132M) modulating binding affinity and thermal stability [7]. Circular dichroism spectroscopy reveals that bilirubin binding enhances the secondary structure content of UnaG variants, contributing to their stability [7].
For metabolite sensing, UnaG derivatives enable highly specific bilirubin detection with dissociation constants in the nanomolar range, allowing development of clinical assays for bilirubin quantification without the need for sample pretreatment [7] [25]. This specificity has been harnessed for developing biosensors for hepatic function and neurological disorders associated with bilirubin metabolism [7].
Table 2: Experimental Performance Comparison
| Parameter | GFP-based Biosensors | Bilirubin-Binding FP Biosensors |
|---|---|---|
| Quantum Efficiency | ~50% (EGFP) | ~50% (UnaG) |
| Excitation/Emission | 488/509 nm (EGFP) | 498/527 nm (UnaG) |
| Brightness Relative to EGFP | 1.0 (reference) | 1.0 (UnaG) |
| Bilirubin Detection Limit | Not applicable | Nanomolar range |
| Monomeric State | Engineered monomers available | Native monomer |
| Oxidative Stress Resistance | Limited | Enhanced [28] |
Experimental Protocols and Methodologies
Expression and Purification of Bilirubin-Binding FPs
The following protocol for UnaG expression and purification has been adapted from published methodologies [7] [5]:
- Vector Construction: Clone the UnaG coding sequence into pColdI expression vector with an N-terminal 6xHis tag using BamHI and EcoRI restriction sites.
- Transformation: Introduce the constructed plasmid into BL21 chemically competent E. coli cells.
- Protein Expression: Grow transformed cells in lysogeny broth with ampicillin (100 μg/mL) at 37°C for 3 hours, then induce with 0.5 mM IPTG and continue growth at 16°C for 18 hours.
- Cell Harvesting: Collect cells by centrifugation, wash with potassium phosphate buffer (20 mM, pH 7.5), and store cell pellets at -80°C.
- Protein Purification: Thaw cells and homogenize by sonication in potassium phosphate buffer. Apply soluble fraction to HisTrap HP column, wash with buffer containing 20 mM imidazole, and elute with elution buffer containing 500 mM imidazole.
- Buffer Exchange: Desalt eluted protein using PD-10 column with potassium phosphate buffer containing 0.02% sodium azide.
For bilirubin-binding assays, prepare a 1 mM bilirubin stock solution in DMSO and store at -20°C protected from light [5].
Bilirubin Binding and Fluorescence Characterization
- Sample Preparation: Mix purified apoprotein with increasing concentrations of bilirubin (0-500 nM) in potassium phosphate buffer.
- Spectroscopic Analysis:
- Record UV-VIS absorption spectra from 300-600 nm to detect characteristic bilirubin absorption peaks upon binding.
- Measure fluorescence excitation and emission spectra using a fluorescence spectrophotometer.
- For kinetic studies, monitor fluorescence intensity at 527 nm following excitation at 498 nm over time.
- Determination of Binding Affinity:
- Plot fluorescence intensity versus bilirubin concentration.
- Fit data to a hyperbolic binding equation to determine dissociation constant (Kd).
- Alternatively, use nonlinear regression analysis for more complex binding models accounting for the two fluorescence states [5].
Diagram 2: Experimental workflow for developing bilirubin-binding FP biosensors.
Biosensor Engineering and Applications
Engineering Strategies for Enhanced Performance
Protein engineering approaches have significantly advanced both GFP-based and bilirubin-binding FP biosensors. For UnaG, site-directed mutagenesis has targeted key residues involved in bilirubin binding [7]. Replacing arginine residues at positions 112 and 132 with methionine reduces polarity at bilirubin-binding sites, altering spectroscopic properties and binding affinity [7]. The development of eUnaG through valine substitution at position 2 demonstrates approximately twofold enhanced fluorescence intensity compared to wild-type UnaG [7].
Structural studies have been instrumental in these engineering efforts. The conserved Gly-Pro-Pro motif in fluorescent FABPs serves as a critical structural element, with mutation to Gly-Gly-Gly significantly reducing quantum yield and deletion fully quenching fluorescence [6] [27]. Residues adjacent to this motif show evidence of positive selection, suggesting ongoing refinement of fluorescent properties in natural systems [27].
Applications in Metabolic Monitoring and Disease Research
Bilirubin-binding FPs have enabled innovative applications in biomedical research:
- Hypoxia Imaging: The oxygen-independent fluorescence of UnaG has been exploited to visualize hypoxic regions in tumors at cellular resolution, overcoming a significant limitation of GFP-based sensors [25].
- Bilirubin Distribution Mapping: UnaG serves as a fluorescent probe for intracellular bilirubin distribution, providing insights into heme catabolism and bilirubin-related pathologies [5] [25].
- Protein-Protein Interaction Studies: Split UnaG systems (uPPI) enable bimolecular fluorescence complementation assays for monitoring protein interactions in living cells [25].
- Dual-Ligand Sensors: Engineering UnaG with calmodulin creates a dual-ligand modulable fluorescent protein that responds to both bilirubin and calcium concentrations, demonstrating the modularity of these biosensors [25].
- Oxidative Stress Resistance: Cells expressing eel fluorescent protein show approximately twofold greater resistance to oxidative stress like H₂O₂ exposure, suggesting protective functions [28].
Research Reagent Solutions
Table 3: Essential Research Reagents for Biosensor Development
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Expression Vectors | pColdI, pGEX, pTolT | Recombinant protein expression in E. coli |
| Chromophore Ligands | Bilirubin (BR), Unconjugated Bilirubin | Fluorogen for bilirubin-binding FPs |
| Chromatography Media | HisTrap HP, Ni-NTA | Affinity purification of tagged proteins |
| Site-Directed Mutagenesis Kits | Phusion, KOD-Plus-Mutagenesis | Protein engineering and optimization |
| Spectroscopic Equipment | Fluorescence spectrophotometers, CD spectroscopy | Characterization of optical properties |
| Cell Culture Reagents | HEK293 cell line, hypoxic chambers | Cellular biosensor validation |
The expanding toolbox of genetically encoded biosensors continues to transform our ability to monitor cellular metabolism and signaling events. While GFP-based biosensors offer a well-established platform with diverse spectral variants, bilirubin-binding FPs provide unique advantages for specific applications, particularly oxygen-independent imaging, metabolite sensing, and hypoxic environment monitoring. The small size, native monomeric state, and instantaneous activation of bilirubin-binding FPs make them valuable complements to traditional GFP systems.
Future developments will likely focus on engineering enhanced variants with improved brightness and photostability, expanding the color palette through structural modifications, and developing multi-analyte sensors that simultaneously monitor multiple metabolic parameters. The continued discovery and characterization of new bilirubin-binding FPs from diverse eel species [6] [27] promises to provide additional templates for biosensor engineering. As these technologies mature, they will undoubtedly yield increasingly sophisticated tools for fundamental research and drug development, enhancing our understanding of cellular metabolism in health and disease.
Fluorescent proteins (FPs) have revolutionized live-cell imaging, transforming our ability to visualize dynamic processes within living systems. As microscopy technologies, particularly super-resolution techniques, advance toward capturing finer structural details at faster speeds, the demand has intensified for FPs with optimized properties. This guide focuses on two distinct families of fluorescent probes: the traditional Green Fluorescent Protein (GFP) and its variants, and the emerging class of bilirubin-inducible fluorescent proteins (FPs). While GFP-like proteins dominate many biological imaging applications, bilirubin-binding FPs like UnaG and its homologs offer unique advantages for specialized imaging contexts, including oxygen-independent maturation and operation in hypoxic environments [29] [5] [19]. The choice between these protein families significantly impacts experimental outcomes in super-resolution and multiparameter imaging, influencing factors from image brightness to temporal resolution and viability in sensitive cellular conditions. This comparison guide provides an objective analysis of their performance characteristics, supported by experimental data, to inform researchers, scientists, and drug development professionals in selecting the optimal probes for their specific imaging challenges.
Technology Comparison: GFP-vs Bilirubin-Binding FPs
Core Mechanisms and Structures
GFP and its Variants: GFP-like proteins feature a beta-barrel structure that encapsulates a chromophore formed via autocatalytic post-translational modification of three internal amino acids. This maturation process requires molecular oxygen, which can limit their use in anaerobic or hypoxic conditions [16] [19]. Engineering efforts have produced enhanced variants like sfGFP (superfolder GFP) and the recently developed YuzuFP, which demonstrates improved brightness and photostability through a single H148S mutation that enhances chromophore interaction [16].
Bilirubin-Binding FPs (UnaG Family): This class belongs to the fatty acid-binding protein family and features a beta-barrel structure that binds bilirubin (BR) as an exogenous chromophore [5] [6]. Unlike GFP, fluorescence is instantaneous and oxygen-independent, as the chromophore does not require oxidative maturation [5]. The UnaG-BR complex exists in two distinct fluorescence states (holoUnaG1 and holoUnaG2) with a reversible transition between them, offering unique photophysical properties [5].
Quantitative Performance Metrics
Table 1: Performance Characteristics of Representative Fluorescent Proteins
| Protein | Class | Excitation (nm) | Emission (nm) | Quantum Yield | Brightness (Relative) | Maturation | Oxygen Requirement |
|---|---|---|---|---|---|---|---|
| sfGFP [16] | GFP variant | 485 | 510 | ~0.65 | 1.0 (reference) | Hours | Yes |
| YuzuFP [16] | GFP variant (H148S) | ~485 | ~510 | Higher than sfGFP | 1.5x sfGFP | Hours | Yes |
| UnaG [5] | Bilirubin-binding | 496 | 527 | ~0.50 | Comparable to EGFP | Instantaneous | No |
| HoloUnaG2 [5] | Bilirubin-binding | ~496 | ~527 | - | 3.9x HoloUnaG1 | Instantaneous | No |
| miniGFP1/2 [19] | Flavin-binding | 450 | 499 | - | >2x parental | Instantaneous | No |
Table 2: Photostability and Advanced Application Performance
| Protein | Class | Photobleaching Resistance | Monomeric | Unique Applications | Key Advantages |
|---|---|---|---|---|---|
| YuzuFP [16] | GFP variant | ~3x sfGFP | Yes | General super-resolution imaging | Enhanced brightness & stability |
| UnaG [5] | Bilirubin-binding | Moderate | Yes [5] | Hypoxic imaging, BR quantification | Oxygen-independent, no maturation delay |
| miniGFP1/2 [19] | Flavin-binding | Improved vs other FbFPs | Yes | Anaerobic imaging, metal sensing | Small size, Cu(II) sensitivity (Kd ~67 nM) |
Distinct Operational Advantages
GFP Variants excel in general-purpose applications where oxygen is available and maturation time is not critical. Their extensive optimization history provides researchers with a wide range of well-characterized tools for most standard imaging scenarios [16].
Bilirubin-Binding FPs offer crucial advantages for specialized imaging contexts: Their oxygen-independent fluorescence enables imaging in hypoxic environments and anaerobic organisms [19]. Instantaneous fluorogen activation allows real-time observation of rapid biological processes without maturation delays [5]. They also function as biosensors for bilirubin, with applications in quantifying heme catabolism in neurological research [30].
Experimental Protocols and Methodologies
Protocol 1: Characterizing Bilirubin-Binding FP Dynamics
The discovery of two distinct fluorescence states in UnaG required sophisticated biophysical characterization [5]:
Protein Purification: Recombinant 6×His-tagged UnaG was expressed in E. coli and purified using immobilized metal affinity chromatography (IMAC) with Ni-NTA resin, followed by desalting into potassium phosphate buffer [5].
Complex Formation: UnaG was mixed with bilirubin (dissolved in DMSO) at specific molar ratios to form the holoUnaG complex [5].
Spectroscopic Analysis: Fluorescence spectra and time-course measurements were performed using a spectrophotometer (RF-5300PC). The team employed photon number counting analysis to quantify the molecular brightness ratio and equilibrium population ratio between holoUnaG1 and holoUnaG2 states [5].
Analytical Ultracentrifugation (AUC): Both sedimentation velocity (AUC-SV) and sedimentation equilibrium (AUC-SE) analyses were conducted to determine the oligomeric state of apoUnaG and holoUnaG under various conditions [5].
Experimental Workflow for UnaG State Characterization
Protocol 2: Engineering Enhanced GFP Variants
The development of YuzuFP from sfGFP employed computational and molecular biology techniques [16]:
Molecular Dynamics Modeling: Short time-scale (10 ns) MD simulations sampled all 19 canonical amino acids at position H148 to predict changes in chromophore interaction networks and solvation [16].
Mutant Construction: Selected mutations (H148S, H148A, H148C, H148N, H148T) were introduced into sfGFP using site-directed mutagenesis [16].
Spectral Characterization: Absorbance and fluorescence spectra of purified mutants were measured to determine excitation/emission maxima and relative brightness [16].
Photostability Assessment: Proteins were subjected to continuous illumination while monitoring fluorescence decay to quantify resistance to photobleaching [16].
Live-Cell Validation: Selected promising variants were expressed in mammalian cells for imaging under realistic conditions to confirm improved performance [16].
Advanced Microscopy Integration
Super-Resolution Imaging with 3D-MP-SIM
Recent advances in structured illumination microscopy (SIM) have created new demands and opportunities for fluorescent proteins. The development of 3D multiplane SIM (3D-MP-SIM) achieves approximately eightfold increased temporal resolution for volumetric super-resolution imaging, with lateral and axial resolutions of approximately 120 nm and 300 nm, respectively [31]. This technology enables high-speed time-lapse volumetric imaging at rates up to 11 volumes per second, ideal for capturing dynamic processes in live cells [31].
For such applications, the photostability of FPs becomes critically important. YuzuFP's 3-fold increased resistance to photobleaching compared to sfGFP makes it particularly valuable for extended super-resolution time-lapse experiments [16]. Similarly, the oxygen-independent fluorescence of bilirubin-binding FPs provides advantages when imaging hypoxic cellular regions or anaerobic organisms at high resolution [19].
Super-Resolution Microscopy Evolution
Multiparameter Imaging Applications
The distinct spectral and functional properties of different FP classes enable sophisticated multiparameter imaging experiments:
Dual-Color Organelle Interaction Studies: 3D-MP-SIM has demonstrated feasibility for dual-color imaging, enabling observation of rapid interactions between intracellular organelles in 3D space [31]. This requires FPs with precspectral separation and minimal cross-talk.
Metabolic and Ionic Sensing: Flavin-binding miniGFPs demonstrate sensitivity to copper ions (Kd ~67 nM for Cu(II)) and the ability to bind flavin mononucleotide, functioning as multisensing platforms for various analytes [19].
Neurological Biomarker Quantification: The HUG (HELP-UnaG) assay enables nanomolar-range detection of bilirubin and biliverdin in cerebrospinal fluid, providing insights into brain heme catabolism relevant to neurodegenerative diseases [30].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Fluorescent Protein Research and Applications
| Reagent/Method | Function | Example Use |
|---|---|---|
| HUG Assay System [30] | Quantify bilirubin/biliverdin at nM concentrations | Measurement of heme catabolism biomarkers in CSF |
| Analytical Ultracentrifugation [5] | Determine oligomeric state and molecular mass | Verification of UnaG monomeric state |
| Photon Counting Analysis [5] | Quantify molecular brightness ratios | Distinguishing holoUnaG1 vs holoUnaG2 states |
| 3D-MP-SIM Microscopy [31] | High-speed volumetric super-resolution imaging | Capturing ER dynamics at 11 volumes/second |
| Directed Molecular Evolution [19] | Protein engineering through iterative screening | Development of miniGFP1/2 from phiLOV3 template |
| Molecular Dynamics Simulations [16] | Predict chromophore-residue interactions | Guiding H148S mutation in YuzuFP development |
The comparative analysis of GFP-like proteins versus bilirubin-binding fluorescent proteins reveals a complementary relationship rather than a competitive one in advanced microscopy applications. GFP variants like YuzuFP currently dominate general super-resolution imaging where their enhanced brightness and photostability provide practical advantages [16]. Meanwhile, bilirubin-binding proteins offer specialized capabilities for hypoxic imaging, anaerobic studies, and metabolic sensing [5] [30] [19].
Future developments will likely focus on engineering bridges between these protein families, creating variants that combine the beneficial properties of each. The successful engineering of YuzuFP through molecular dynamics-guided single mutation demonstrates the potential for rational design to enhance FP performance [16]. Similarly, the discovery of distinct fluorescence states in UnaG opens possibilities for developing brighter mutants constitutively stabilized in the holoUnaG2 state [5].
As microscopy technologies continue advancing toward faster volumetric imaging and higher resolution [31], both protein families will evolve to meet these demands. The ideal FP of the future may combine the oxygen-independent maturation of bilirubin-binding proteins with the exceptional brightness and photostability of optimized GFP variants, potentially through novel engineering approaches or the discovery of entirely new FP classes from unexplored biological sources [6].
The development of cell and gene therapies, such as chimeric antigen receptor (CAR) T cells and viral gene transfer vectors, represents a breakthrough in treating life-threatening diseases. However, the clinical translation of these advanced therapy medicinal products (ATMPs) remains hampered by limited knowledge about their biodistribution, persistence, and dynamic behavior in living organisms. Technologies to non-invasively and quantitatively monitor the distribution of ATMPs in vivo could greatly improve our understanding of their trafficking, therapeutic efficacy, and off-target toxicity. Likewise, adeno-associated virus (AAV)-based gene therapies would benefit from quantifying the location, magnitude, and duration of transgene expression. This guide provides a comprehensive comparison of current technologies for tracking therapeutic cells and viral vectors in vivo, with particular emphasis on fluorescent protein applications within a broader comparison framework of GFP versus alternative imaging modalities.
Comparative Analysis of In Vivo Tracking Technologies
The table below summarizes the key characteristics, advantages, and limitations of major technologies used for tracking therapeutic cells and viral vectors in vivo.
Table 1: Comparison of Major In Vivo Tracking Technologies
| Technology | Spatial Resolution | Detection Sensitivity | Temporal Tracking Capacity | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| PET with Reporter Genes [32] | 1-2 mm (preclinical) | ~1,200 CAR T cells [32] | Longitudinal (≥4 weeks) [32] | Whole-body, quantitative, depth-independent, high sensitivity [32] | Requires reporter introduction, radiation exposure, complex radiochemistry |
| Super-resolution Microscopy (dSTORM) [33] | Single-molecule (~20 nm) [33] | Single CAR detection [33] | Terminal (ex vivo) | Tag-free CAR detection, single-molecule sensitivity, quantifies CAR density [33] | No in vivo capability, requires specialized equipment and expertise |
| Magnetic Particle Imaging (MPI) [34] | N/A (in search results) | Ultra-sensitive and linear quantitative imaging [34] | Longitudinal (demonstrated ≥7 days) [34] | Near-ideal contrast, penetration depth, safety (non-ionizing) [34] | Limited clinical translation to date, primarily preclinical |
| Fluorescence Imaging (GFP-like proteins) [35] | Limited by light scattering | Limited by tissue depth | Varies with protein stability | Self-contained fluorescence, no cofactors, broad fusion compatibility [35] | Poor tissue penetration, autofluorescence interference, photostability issues [34] |
| Magnetic Resonance Imaging (MRI) [34] | High (~50 μm) [34] | Varies with contrast agent | Longitudinal (demonstrated ≥2 weeks) [34] | Detailed anatomical context, clinically established, non-ionizing [34] | Lower sensitivity versus nuclear imaging, potential contrast agent toxicity |
Detailed Experimental Protocols for Key Tracking Methodologies
PET Reporter System Based on Membrane-Anchored Anticalin Protein
Experimental Workflow:
- Reporter Design: Construct a fusion protein comprising the lipocalin-2 signal peptide, an engineered anticalin binding protein, a peptide linker containing a V5-tag, and the human CD4 transmembrane domain (total 257 amino acids) [32].
- Cell Engineering: Introduce the reporter gene into therapeutic cells (CAR T cells or AAV-transduced cells) using retroviral or lentiviral vectors [32].
- Radioligand Preparation: Synthesize the cognate reporter probe featuring CHX-A″-DTPA•metal or colchicine binding moiety, a PEG4-linker, and a labeling group for efficient incorporation of the fluorine-18 isotope [32].
- In Vivo Imaging: Administer the 18F-labelled radioligand intravenously and perform longitudinal PET imaging over weeks to monitor cell expansion and migration [32].
- Validation: Correlate PET signal intensity with flow cytometry data and ex vivo analyses to validate quantitative accuracy [32].
Direct CAR Visualization Using dSTORM Super-Resolution Microscopy
Experimental Workflow:
- CAR-T Cell Generation: Isolate human PBMCs from healthy donors and transduce with lentiviral vectors encoding CAR constructs with optional EGFRt transduction marker [33].
- Sample Preparation: Fix CAR-T cells on appropriate microscopy slides and permeabilize if needed for intracellular targets [33].
- Immunostaining: Incubate cells with primary antibody specific for CAR backbone (e.g., anti-IgG4 F(ab)2 fragment) followed by dye-conjugated secondary antibodies (e.g., Alexa Fluor 647) [33].
- dSTORM Imaging: Acquire images in a photoswitching buffer under total internal reflection fluorescence (TIRF) microscopy conditions. Collect 10,000-30,000 frames at high laser power to induce stochastic blinking [33].
- Image Reconstruction and Analysis: Localize single molecules with nanometer precision and reconstruct super-resolution images. Quantify CAR surface density and spatial organization using cluster analysis algorithms [33].
Quantitative Performance Data for Tracking Technologies
Table 2: Quantitative Performance Metrics of Tracking Technologies
| Technology | Quantitative Performance Metrics | Experimental Context | Temporal Resolution |
|---|---|---|---|
| PET Reporter System [32] | >800-fold higher radioligand binding vs controls; Detection of 1,200 CAR T cells in bone marrow; Linear correlation with flow cytometry (R² not specified) [32] | Mouse models of CAR T cell therapy and AAV gene transfer | Longitudinal monitoring over 4 weeks [32] |
| Super-resolution Microscopy [33] | Single-molecule sensitivity; CAR density quantification; Identification of CAR clusters/µm² [33] | Primary human CAR-T cells targeting SLAMF7, BCMA, CD19 | Endpoint analysis (single time point) |
| Fluorine-19 MRI [35] | Detection of 19F perfluorocarbon labeled CAR-T cells for up to 7 days post-injection without significant impact on cytotoxicity [35] | Mouse models of leukemia | Up to 7 days post-injection [35] |
| Ferumoxytol-labeled MRI [34] | Significant T2 shortening: 22.2 ms ± 3.2 vs 27.9 ms ± 1.8 in unlabeled controls (P < 0.001); Higher T2 in apoptotic vs viable implants (26.6 ms ± 4.9 vs 20.8 ms ± 5.3; P = 0.001) [34] | Minipig model of cartilage repair | Early diagnosis of failed implants within 2 weeks [34] |
| Bioluminescence (AkaBLI) [34] | 100-1000-fold brighter signals compared to conventional technology; Recording from deep brain neurons for >1 year [34] | Mouse striatum neurons | Over 1 year continuous monitoring [34] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Research Reagents for Tracking Therapeutic Cells
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Reporter Proteins | Engineered anticalins (DTPA-R, Colchi-R) [32]; GFP variants (mHoneydew, mBanana, mOrange, tdTomato, mCherry) [35] | Enable detection via binding to specific ligands or intrinsic fluorescence |
| Viral Vectors | Lentiviral vectors (VSV-G pseudotyped, CD8-targeted, CD4-targeted) [36] [33]; γ-retroviral vectors [37] | Efficient gene delivery to therapeutic cells |
| Detection Probes | 18F-labelled lanthanide complex [32]; Anti-IgG4 F(ab)2 fragments [33]; Fluorophore-conjugated antigens [35] | Direct visualization and quantification of labeled cells |
| Transduction Enhancers | Rapamycin [36]; Engineered envelope proteins [36] | Improve gene transfer rates by downregulating antiviral restriction factors |
| Contrast Agents | Superparamagnetic iron oxide nanoparticles (SPIONs) [34]; 19F perfluorocarbons [35]; Ferumoxytol [34] | Enable MRI and MPI tracking of labeled cells |
Integration with Broader Thesis: GFP vs. Bilirubin-Binding Fluorescent Proteins
While GFP and its variants (mFruits series including mCherry, tdTomato, mStrawberry) have been valuable tools for validating CAR expression [35], they face limitations including poor photostability, susceptibility to enzymatic degradation, and interference from biosubstrate autofluorescence [34]. These limitations are particularly problematic for long-term in vivo tracking where signal stability and depth penetration are crucial.
The search results highlight a shift toward more specialized technologies that address these limitations. PET reporter systems offer quantitative, depth-independent whole-body imaging [32], while super-resolution microscopy provides unprecedented spatial resolution at the single-molecule level, albeit limited to ex vivo applications [33]. The development of brighter bioluminescence systems (AkaBLI) with 100-1000-fold enhanced signals demonstrates ongoing innovation in optical imaging that may bridge the sensitivity gap between traditional fluorescence and nuclear imaging approaches [34].
Each tracking modality presents distinct advantages depending on the research question: PET for quantitative whole-body biodistribution, super-resolution microscopy for detailed mechanistic studies of CAR organization, and advanced bioluminescence for long-term monitoring of cellular processes in deep tissues. The optimal choice depends on the specific requirements for spatial resolution, temporal tracking, quantification needs, and whether in vivo or ex vivo analysis is required.
The field of therapeutic cell tracking has evolved significantly beyond conventional fluorescent proteins, with current technologies enabling unprecedented sensitivity and resolution. PET reporter systems provide exceptional sensitivity for in vivo quantification, while super-resolution microscopy offers nanometer-scale resolution for detailed mechanistic studies. The choice of tracking technology should be guided by specific research objectives, considering the trade-offs between spatial resolution, temporal tracking capability, and detection sensitivity. As these technologies continue to advance, they will provide increasingly sophisticated insights into the behavior and mechanisms of action of CAR-T cells and viral gene transfer vectors in living organisms.
Solving Research Challenges: A Troubleshooting Guide for Fluorescent Tags
Fluorescent proteins have revolutionized molecular imaging, yet their application is constrained by fundamental physiological barriers. This guide provides a comparative analysis of green fluorescent protein (GFP) and bilirubin-binding fluorescent proteins, focusing on their performance under anaerobic conditions. We detail how bilirubin-binding proteins, such as UnaG, overcome the critical oxygen dependency of GFP, enabling live-cell imaging in low-oxygen environments like hypoxic tumors and anaerobic microbial communities. Structured experimental data and protocols are provided to equip researchers with practical knowledge for selecting appropriate fluorescent reporters for their specific biological contexts.
The discovery and development of the Green Fluorescent Protein (GFP) and its variants represent a cornerstone of modern biological imaging, enabling researchers to visualize dynamic cellular processes in real-time [38]. These proteins have become indispensable tools for tracking gene expression, protein localization, and protein-protein interactions in living systems [39]. However, a significant limitation inherent to GFP and related analogs derived from jellyfish and corals is their strict requirement for molecular oxygen to develop fluorescence [40] [20].
The maturation process of GFP involves an intramolecular reaction that forms the chromophore, a process that is fundamentally dependent on the presence of oxygen [20]. This dependency makes GFP unreliable and often completely non-functional in low-oxygen or anaerobic environments, severely limiting its application for studying biological systems that thrive in such conditions [40]. These systems include gut microbiota (which are predominantly anaerobic), hypoxic tumor microenvironments, and various microbial communities central to biotechnological applications like sustainable biomanufacturing [40].
In response to these limitations, alternative fluorescent protein systems that operate independently of oxygen have been developed. Among these, bilirubin-binding fluorescent proteins, notably UnaG, represent a powerful class of oxygen-independent reporters [40] [20]. This guide provides a comprehensive comparison of the performance characteristics of GFP versus bilirubin-binding proteins, with a specific focus on their application in anaerobic conditions, supported by experimental data and detailed protocols for their use.
Performance Comparison: GFP vs. Bilirubin-Binding Proteins
The table below summarizes the key characteristics of GFP and bilirubin-binding fluorescent proteins, highlighting critical differences that impact their performance in various research contexts.
Table 1: Performance Comparison of GFP and Bilirubin-Binding Fluorescent Proteins
| Characteristic | GFP and Derivatives | Bilirubin-Binding Proteins (e.g., UnaG) |
|---|---|---|
| Oxygen Requirement | Required for chromophore maturation [40] [20] | Not required [40] |
| Chromophore Origin | Autocatalytic from internal amino acids [38] | Binds bilirubin, a heme metabolite [40] |
| Primary Binding Site | N/A (internal chromophore) | Subdomain IB of human serum albumin [41] |
| Molecular Size | ~25-28 kDa [38] [20] | ~13-16 kDa (smaller size) [40] |
| Key Applications | General live-cell imaging in aerobic conditions [38] | Imaging in anaerobes, hypoxic cells, and deep tissues [40] |
| Major Limitation | Unreliable in anaerobic and hypoxic conditions [40] | Requires bilirubin supplementation in some cell types [40] |
Analytical Performance and Environmental Sensitivity
Beyond the fundamental difference in oxygen requirements, several other performance factors are critical for experimental design.
Table 2: Analytical and Physical Properties Comparison
| Property | GFP and Derivatives | Bilirubin-Binding Proteins |
|---|---|---|
| pH Sensitivity | pKa ~6.0; sensitive to acidic conditions [20] | Stable across a broad pH range [19] |
| Tissue Penetration | Limited (excitation in blue/green spectrum) [40] | Improved (red-shifted spectra variants available) [40] |
| Fluorescence Mechanism | Fluorescence depends on chromophore environment [38] | Fluorescence activated by bilirubin binding [40] |
| Chromophore Supply | Self-sufficient in aerobic cells | Dependent on endogenous bilirubin or exogenous supply [40] |
Experimental Protocols for Anaerobic Imaging
Protocol 1: Validating Fluorescence Under Anaerobic Conditions
This protocol is designed to test and compare the functionality of GFP and bilirubin-binding proteins in a controlled anaerobic environment.
Key Research Reagent Solutions:
- Anaerobic Chamber: Creates and maintains an oxygen-free atmosphere (e.g., with 5% H₂, 5% CO₂, 90% N₂) for cell culture and imaging.
- Fluorescent Protein Constructs: Plasmid DNA or stable cell lines expressing GFP and UnaG.
- Bilirubin Stock Solution: Prepared in a suitable solvent (e.g., DMSO) for supplementing UnaG-expressing cultures.
- Anaerobic Gas Pack System: For generating anaerobic conditions in sealed jars if a chamber is not available.
Methodology:
- Cell Culture and Transfection: Culture appropriate cells (e.g., E. coli or mammalian cells) and transfert with plasmids encoding GFP and UnaG using standard protocols.
- Anaerobic Setup: Divide the cultures into two sets. Place one set in a standard aerobic incubator and the other in an anaerobic chamber or sealed jar with an anaerobic gas pack system.
- Bilirubin Supplementation: For cultures expressing UnaG, add bilirubin to the medium (e.g., 1-10 µM final concentration) to ensure chromophore availability [40].
- Image Acquisition: After 24-48 hours, image the cells. Aerobic samples can be imaged on a standard fluorescence microscope. For anaerobic samples, use a microscope with a stage enclosed within the anaerobic chamber or use sealed imaging dishes to maintain anaerobic conditions during transfer.
- Quantification: Measure the fluorescence intensity of both reporters under both aerobic and anaerobic conditions. Normalize the signals to cell viability or protein expression controls.
Expected Outcome: GFP fluorescence will be significantly diminished or absent under anaerobic conditions, whereas UnaG fluorescence should remain stable, provided bilirubin is available [40] [20].
Protocol 2: Assessing Bilirubin Dependency of UnaG Fluorescence
This protocol determines the necessity of bilirubin supplementation for UnaG fluorescence in different cell types.
Methodology:
- Cell Line Selection: Use a panel of cell lines, including those known to produce bilirubin (e.g., hepatocyte-derived lines) and those that do not (e.g., many bacterial cells).
- Expression: Express UnaG in the selected cell lines.
- Bilirubin Titration: For each cell type, set up cultures with a range of bilirubin concentrations (e.g., 0 nM, 100 nM, 500 nM, 1000 nM) added to the growth medium.
- Flow Cytometry Analysis: After an appropriate expression period, analyze the cells using flow cytometry to measure the fluorescence intensity of the population.
- Kd Determination: Plot fluorescence intensity against bilirubin concentration and fit the data to a binding curve to estimate the apparent Kd for the bilirubin-UnaG interaction in different cellular environments.
Expected Outcome: Fluorescence in cells with endogenous bilirubin production will be less dependent on exogenous supplementation, while cells lacking bilirubin will show a concentration-dependent increase in fluorescence [40].
The Scientist's Toolkit: Essential Research Reagents
The following table outlines key materials required for experiments comparing fluorescent proteins under anaerobic conditions.
Table 3: Essential Research Reagents for Anaerobic Fluorescence Studies
| Reagent / Material | Function & Importance | Example Application |
|---|---|---|
| UnaG Plasmid DNA | Genetically encodes the bilirubin-binding fluorescent protein. | Expression in target cells to enable oxygen-independent reporting. |
| Bilirubin | The chromophore ligand required for UnaG fluorescence. | Supplementation in cell cultures that lack endogenous bilirubin. |
| Anaerobic Chamber | Provides an oxygen-free environment for sample processing and imaging. | Maintaining strict anaerobic conditions for cell culture and fluorescence measurement. |
| Flavin-Binding FPs (e.g., miniGFP) | Alternative oxygen-independent fluorescent proteins that bind endogenous flavins [19]. | Useful as a comparative control or for multi-analyte sensing in anaerobes. |
| Fluorescence Plate Reader / Microscope | Quantifies fluorescence output from samples. | Measuring and comparing the performance of different fluorescent reporters. |
| Anti-GFP Nanobodies | Binds GFP for protein detection in immunoblotting, even when fluorescent signal is lost [15]. | Verifying protein expression in anaerobic samples where GFP fluorescence is absent. |
Experimental Workflow and Signaling Pathways
The diagram below illustrates the fundamental mechanistic difference between GFP and bilirubin-binding proteins, which underpins their performance in anaerobic environments.
Figure 1: Mechanism of Fluorescence in Aerobic vs. Anaerobic Conditions.
The workflow demonstrates that GFP chromophore maturation is blocked in the absence of oxygen, preventing fluorescence. In contrast, the bilirubin-binding protein UnaG fluoresces upon binding bilirubin, a process independent of oxygen availability [40] [20].
The selection of an appropriate fluorescent reporter is paramount for the success of imaging experiments, particularly when studying biological systems that exist in low-oxygen environments. While GFP remains an excellent tool for a wide range of applications under aerobic conditions, its fundamental oxygen dependency renders it unsuitable for anaerobic work.
Bilirubin-binding proteins like UnaG provide a robust, oxygen-independent alternative, enabling researchers to probe previously inaccessible biological spaces, from the human gut microbiome to hypoxic regions of tumors. While considerations such as bilirubin availability and potential spectral overlap must be accounted for in experimental design, the unique advantages of these proteins make them indispensable tools in the expanding molecular imaging toolkit. Future developments in protein engineering will likely yield enhanced bilirubin-binding proteins with improved brightness and spectral diversity, further solidifying their role in anaerobic research.
Fluorescent proteins (FPs) have revolutionized biological imaging, enabling researchers to visualize cellular components and processes with exceptional precision. However, their application in studying acidic organelles—including endosomes, secretory granules, lysosomes, and plant vacuoles (pH ∼4.5–6.0)—has been severely limited by a fundamental constraint: most conventional FPs lose fluorescence in acidic conditions due to their neutral pKa (approximately 6.0) [42]. This sensitivity arises from the protonation of the chromophore's electron-rich, light-absorbing component in sub-physiological pH environments, leading to contracted π conjugation systems and subsequent fluorescence quenching [42] [43]. This limitation has impeded research on critical biological processes such as autophagy, receptor-mediated endocytosis, and lysosomal function.
Within this challenging landscape, two distinct classes of fluorescent proteins have emerged as promising solutions: engineered pH-stable GFP variants and unique bilirubin-binding fluorescent proteins. This guide provides an objective comparison of these protein families, presenting key experimental data to inform selection for acidic environment applications. We evaluate their performance characteristics, operational mechanisms, and practical implementation requirements to empower researchers in making evidence-based decisions for their specific experimental needs.
Understanding Fluorescent Protein Families: GFP Variants vs. Bilirubin-Binding Proteins
Engineered GFP Variants
The green fluorescent protein (GFP) from Aequorea victoria represents the foundational scaffold from which most fluorescent proteins are derived [16]. Traditional GFPs and yellow FPs (YFPs) typically exhibit pH sensitivity with pKa values ranging from 5.7 to 6.0 (e.g., mNeonGreen: pKa 5.7, mVenus: pKa 6.0) [42]. This sensitivity has driven extensive protein engineering efforts to develop acid-tolerant variants through rational design and directed evolution approaches.
Table 1: Acid-Tolerant Engineered GFP Variants and Their Properties
| Protein | λex/em (nm) | Brightness | pKa | Oligomeric State | Key Features |
|---|---|---|---|---|---|
| Gamillus | 504/519 | 74.7 | 3.4 | Monomer | Excellent brightness, maturation speed, and photostability in acidic conditions [42] [44] |
| pH-tdGFP | 488/515 | N/D | 4.8 | Tandem dimer | Maintains fluorescence properties in acidic conditions (pH 3.75-8.50) [42] [43] |
| mTagBFP2 | 399/454 | 32.4-36.5 | 2.7-2.4 | Monomer/Oligomer | Blue fluorescent protein with exceptional acid tolerance [42] |
| mCherry | 587/610 | 15.8-25.5 | <4.5-3.8 | Monomer | Red fluorescent protein with good acid tolerance [42] |
| mScarlet | 569/594 | 70.0 | 5.3 | Monomer | Bright red fluorescent protein with moderate acid tolerance [42] |
| Sirius | 355/424 | 3.6 | <3.0 | Monomer | Blue fluorescent protein with excellent acid tolerance [42] |
| mTurquoise2 | 434/474 | 27.9-28.5 | 3.1-3.6 | Monomer | Cyan fluorescent protein with good acid tolerance [42] |
Notably, Gamillus, cloned from the flower hat jellyfish (Olindias formosa), represents a significant advancement with its exceptionally low pKa of 3.4 and nearly twice the brightness of previously reported GFPs, maintaining constant fluorescence spectrum between pH 4.5 and 9.0 [44]. The molecular basis for Gamillus's acid tolerance involves stabilization of deprotonation in its chemical structure, as revealed through X-ray crystallography and point mutagenesis studies [44].
Bilirubin-Binding Fluorescent Proteins
In contrast to GFP-derived proteins, an entirely different class of fluorescent proteins has been discovered in eels and other marine organisms. These proteins belong to the fatty-acid binding protein (FABP) family and exhibit unique ligand-induced fluorescence mechanisms. Unlike conventional FPs that generate fluorescence through autocatalytic chromophore formation, these proteins require binding with bilirubin (a linear tetrapyrrole chromophore produced by heme catabolism) to become fluorescent [5] [6] [7].
Table 2: Bilirubin-Binding Fluorescent Proteins and Their Properties
| Protein | Source Organism | λex/em (nm) | Molecular Weight (kDa) | Key Features |
|---|---|---|---|---|
| UnaG | Japanese eel (Anguilla japonica) | 498/527 | 15.6 | First discovered bilirubin-inducible FP; high quantum efficiency (~50%) [5] [7] |
| GymFP | Moray eel (Gymnothorax zonipectis) | 496/532 | 15.6 | 61% homologous to UnaG; extends FABP FPs to third family of true eels [6] |
| Chlopsid FP I | Kaupichthys eels | N/D | N/D | Contains conserved Glycine-Proline-Proline (GPP) motif essential for fluorescence [6] |
The UnaG protein exemplifies this class, functioning as a homodimer that binds bilirubin non-covalently within its β-barrel structure [5]. In its apo state (apoUnaG), the protein remains non-fluorescent until it specifically binds bilirubin to form the fluorescent holoUnaG complex [5]. This unique mechanism provides inherent advantages for acidic environments, as the fluorescence depends on ligand binding rather than chromophore protonation states sensitive to pH.
Direct Comparison: Performance in Acidic Environments
Quantitative Performance Metrics
Table 3: Comprehensive Performance Comparison in Acidic Conditions
| Parameter | Engineered GFP Variants | Bilirubin-Binding Proteins |
|---|---|---|
| Acid Tolerance | pKa as low as 3.4 (Gamillus) [42] [44] | Function independently of chromophore protonation states [5] |
| Brightness | Gamillus: 74.7; mScarlet: 70.0 [42] | UnaG: Quantum efficiency ~50% (equivalent to EGFP) [5] |
| Maturation | Rapid, oxygen-dependent (hours) [16] | Instantaneous, oxygen-independent [5] |
| Photostability | Varies; Gamillus exhibits excellent photostability [44] | Potentially enhanced via ligand exchange [5] |
| Signal Stability | Stable fluorescence across pH 4.5-9.0 (Gamillus) [44] | Two distinct fluorescence states with reversible transition [5] |
| Applications | General organelle labeling; long-term imaging [42] [44] | BR sensing; hypoxic imaging; targeted applications [5] [7] |
Molecular Mechanisms of Acid Tolerance
The fundamental difference in how these protein families achieve acid tolerance stems from their distinct fluorescence mechanisms. For conventional GFPs, acid quenching occurs through protonation of the chromophore phenolate oxide in acidic conditions. This protonation contracts the π conjugation system, shifting absorption to the UV spectrum and increasing thermal relaxation that diminishes fluorescence quantum yield [42].
Engineered acid-tolerant GFPs address this limitation through structural modifications that stabilize the deprotonated state. In Gamillus, specific residue arrangements and hydrogen bonding networks stabilize the chromophore against protonation [44]. Similarly, pH-tdGFP incorporates N149Y and Q204H mutations that significantly enhance pH stability, though these mutations incidentally promote dimerization, necessitating a tandem dimer construct for practical application [43].
In contrast, bilirubin-binding proteins like UnaG completely bypass the protonation sensitivity issue. Their fluorescence derives from the bilirubin ligand itself, protected within the protein's β-barrel structure. The conserved Glycine-Proline-Proline (GPP) motif found in these proteins resides on a protective loop that shields bilirubin from solvent, contributing to fluorescence stability across pH variations [6]. This unique mechanism explains why UnaG maintains high quantum efficiency (~50%) comparable to enhanced GFP, despite pH fluctuations [5].
Experimental Protocols for Characterization
Standardized Acid Tolerance Assessment
Protocol 1: pH Titration for Fluorescence Stability Measurement
- Protein Purification: Express and purify fluorescent proteins using standard systems (e.g., E. coli expression with His-tag purification) [5] [43].
- Buffer Preparation: Prepare buffers across pH range 3.75-8.50, maintaining constant ionic strength [43].
- Sample Incubation: Incubate purified proteins in each buffer condition for stabilization.
- Spectral Acquisition: Measure excitation and emission spectra at each pH using fluorescence spectrophotometry [43].
- Data Analysis: Calculate pKa by fitting fluorescence intensity vs. pH to appropriate models [43].
- Brightness Calculation: Determine brightness as product of molar extinction coefficient and quantum yield [42].
Protocol 2: Live-Cell Acidic Organelle Imaging
- Construct Design: Create fusion proteins targeting acidic organelles (e.g., LAMP1-Gamillus for lysosomes) [44].
- Cell Transfection: Introduce constructs into appropriate cell lines using standard transfection methods.
- pH Manipulation: Apply compounds like bafilomycin A1 to manipulate organelle pH as experimental control [42].
- Confocal Imaging: Image using standard laser lines and filters compatible with FP spectra [45] [44].
- Quantitative Analysis: Measure fluorescence intensity over time to assess photostability under acidic conditions.
Specialized Characterization for Bilirubin-Binding Proteins
Protocol 3: Bilirubin Binding Affinity Measurement
- Protein Preparation: Express and purify apoUnaG or related proteins [5] [7].
- Ligand Titration: Titrate bilirubin (dissolved in DMSO) into protein solutions.
- Fluorescence Monitoring: Measure fluorescence intensity increase upon bilirubin binding [5].
- KD Calculation: Determine dissociation constants by fitting binding isotherms [7].
- Structural Analysis: Employ X-ray crystallography or molecular dynamics to characterize binding interactions [5] [7].
Experimental Workflow for Characterizing pH-Resistant Fluorescent Proteins
Mechanism Visualization: Fluorescence in Acidic Environments
Molecular Mechanisms of Acid Tolerance in Fluorescent Protein Families
Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for pH-Stable FP Applications
| Reagent/Material | Function | Example Applications |
|---|---|---|
| Gamillus plasmid | Acid-tolerant GFP variant | Lysosomal/endosomal labeling; autophagy studies [44] |
| pH-tdGFP construct | Tandem dimer GFP with pH stability | Quantitative imaging in acidic organelles [43] |
| UnaG expression system | Bilirubin-inducible FP | BR detection; hypoxia sensing; specialized imaging [5] [7] |
| Bilirubin standard | Ligand for UnaG activation | Fluorescence activation; binding affinity measurements [5] [7] |
| Organelle markers | Specific compartment labeling | Co-localization studies; compartment-specific pH effects [42] |
| pH calibration buffers | Standardized pH conditions | pH titration experiments; quantitative stability assessment [43] |
| Protease inhibitors | Sample preservation | Maintaining protein integrity during purification [42] |
Application Guidelines and Selection Framework
Protein Selection Decision Matrix
When selecting appropriate pH-resistant variants for specific research applications, consider the following decision framework:
For General Acidic Organelle Labeling: Gamillus provides an optimal balance of brightness, acid tolerance (pKa 3.4), and monomeric behavior, making it suitable for most lysosomal, endosomal, and autophagosomal labeling applications [42] [44].
For Quantitative Imaging Experiments: pH-tdGFP offers exceptional fluorescence stability across broad pH ranges (3.75-8.50), making it ideal for quantitative measurements where signal consistency is critical [43].
For Bilirubin Sensing and Metabolic Studies: UnaG and related bilirubin-binding proteins provide specific detection capabilities for bilirubin distribution, concentration monitoring, and heme metabolism studies [5] [7].
For Multicolor Imaging in Acidic Environments: Combine Gamillus (green) with acid-tolerant red FPs like mCherry (pKa <4.5) or mScarlet (pKa 5.3) for dual-color experiments in acidic compartments [42].
For Hypoxic or Anaerobic Conditions: Bilirubin-binding proteins offer advantages due to their oxygen-independent fluorescence activation, unlike traditional GFPs that require oxygen for chromophore maturation [5].
Implementation Considerations
Successful implementation of pH-resistant fluorescent proteins requires attention to several practical considerations. For engineered GFP variants, ensure proper folding and maturation by maintaining appropriate expression conditions, as some mutations enhancing acid tolerance may impact expression efficiency [43] [44]. For bilirubin-binding proteins, supplement with bilirubin as needed and account for potential effects on cellular processes due to bilirubin manipulation [5] [7]. Always include appropriate controls for pH variation when interpreting results, and validate localization with compartment-specific markers, particularly when studying unfamiliar acidic organelles [42].
The continued development of both engineered GFP variants and bilirubin-binding proteins promises enhanced tools for investigating biological processes in acidic environments. Each class offers distinct advantages, with GFP variants generally excelling in universal labeling applications and bilirubin-binding proteins providing specialized functionality for specific research contexts including metabolite sensing and hypoxic condition imaging.
Autofluorescence, the background fluorescence emitted naturally by tissues and fixatives, presents a significant challenge in fluorescence-based research. This inherent signal can obscure the detection of low-abundance analytes and be mistakenly identified as a target of interest, compromising the accuracy of experimental results in fields like drug development and cellular imaging [46]. The struggle is particularly acute when working with model organisms such as C. elegans, where the signal from weakly expressed green fluorescent protein (GFP) fusion proteins is often masked by strong autofluorescence from intestinal granules [47].
This guide objectively compares the performance of traditional GFP with bilirubin-binding fluorescent proteins as tools for mitigating autofluorescence. We will summarize quantitative data in structured tables, provide detailed experimental methodologies, and outline key reagent solutions to help researchers select the optimal approach for their specific tissue imaging applications.
Autofluorescence in biological samples originates from two primary categories: endogenous intracellular components and fixative-induced artifacts. Key endogenous fluorophores include flavins (FAD, FMN), which absorb between 450-500 nm and emit in the blue to green spectrum; reduced nicotinamide adenine dinucleotide (NADH); collagen and elastin in connective tissues; lipofuscin age-pigments; and the heme groups in red blood cells [46] [48]. Aldehyde fixatives like formalin and glutaraldehyde are well-known for generating autofluorescence through the formation of Schiff bases by reacting with amine groups [46].
A critical finding is that cellular autofluorescence is not static; it can increase significantly under stress conditions. Bactericidal treatments, for example, have been shown to induce a substantial increase in green cellular autofluorescence in E. coli, a phenomenon that requires de novo protein synthesis and is evolutionarily conserved across bacterial species, yeast, and human cells [49]. This dynamic nature further complicates the separation of true signal from background.
Head-to-Head Comparison: GFP vs. Bilirubin-Binding FPs
The following table summarizes the core characteristics of Green Fluorescent Proteins and bilirubin-binding fluorescent proteins, highlighting their respective advantages in combating autofluorescence.
Table 1: Characteristics of GFP and Bilirubin-Binding Fluorescent Proteins
| Characteristic | Green Fluorescent Protein (GFP) | Bilirubin-Binding Fluorescent Proteins (e.g., UnaG) |
|---|---|---|
| Chromophore | Intrinsic, formed by post-translational modification (requires O₂) [19] | Bilirubin ligand, incorporated by binding [5] |
| Maturation | Requires oxygen, can take hours [5] | Instantaneous upon bilirubin binding [5] |
| Oxygen Requirement | Required for chromophore formation [19] | Not required [5] |
| Primary Emission Range | Green (e.g., ~509 nm) [47] | Green [5] |
| Key Advantage for Mitigating Autofluorescence | Well-established, many optimized variants; can be separated from autofluorescence using specialized filter sets [47] | Emission can be spectrally separated from common autofluorescence; high quantum efficiency [5] |
| Key Limitation | Signal often overlaps with common autofluorescence (350-550 nm); requires oxygen [46] [47] | Requires the presence of bilirubin; binding kinetics and stability can be complex [5] |
Strategic Approaches for Cleaner Signals
Several effective strategies exist to minimize the impact of autofluorescence, which can be employed individually or in combination. The following diagram illustrates the three primary strategic pathways.
Spectral Separation and Fluorophore Selection
A fundamental tactic is to select fluorophores whose emission is spectrally distinct from the sample's autofluorescence. Since autofluorescence is often most prominent in the blue to green spectrum (350–550 nm), switching to dyes that emit in the red to far-red region (620–750 nm) can dramatically improve the signal-to-background ratio [46]. Bilirubin-binding proteins like UnaG provide a green fluorescence that, while appearing in a crowded spectral region, can be highly efficient and leveraged with other techniques.
Advanced Optical and Image Analysis Techniques
Time-Gated Detection: This powerful method utilizes fluorescent probes with long emission lifetimes, such as azadioxatriangulenium (ADOTA) dyes (~15 ns), which are much longer than the short lifetimes (typically 1-4 ns) of autofluorescence components. By using pulsed excitation and delaying detection by just 10-20 ns, over 96% of the autofluorescence can be eliminated while retaining a significant portion of the probe's signal [48].
Specialized Filter Sets: For GFP imaging, a triple-band filter setup can separate the true GFP signal from autofluorescence. This setup uses a narrow excitation band (e.g., 485/10 nm) and an emission filter with two pass-through bands (e.g., 520/20 nm for GFP and 595/40 nm for autofluorescence), allowing the GFP to appear green and the autofluorescence to appear yellow in a color image [47].
AI-Powered Image Analysis: Software solutions using deep learning classifiers can be trained to recognize and remove areas of autofluorescence from the final analysis area. This is particularly effective for large, structured autofluorescence from sources like red blood cells or collagen [50].
Sample Processing and Chemical Reduction
Chemical Quenching: Treating tissues with chemical agents like Sudan Black B, copper sulfate, or sodium borohydride can effectively quench autofluorescence. The choice of agent can be tissue-specific [46] [50].
Fixation Optimization: Aldehyde fixatives are a major source of autofluorescence. Replacing them with organic solvents like ice-cold ethanol or methanol, or reducing paraformaldehyde concentration and exposure time, can minimize this issue [46].
Removal of Red Blood Cells: The heme groups in red blood cells are a major source of autofluorescence. These can be removed from tissues by perfusion with PBS prior to fixation or from blood samples via lysis and thorough washing [46].
Experimental Protocols for Direct Comparison
To objectively compare the performance of GFP and bilirubin-binding FPs in a high-background context, the following experimental protocols can be employed.
Protocol 1: Evaluating GFP Signal Purity with a Triple-Band Filter Set
This method is adapted from a study on visualizing SKN-1::GFP in C. elegans, whose signal is normally masked by intestinal autofluorescence [47].
Workflow:
- Sample Preparation: Use transgenic C. elegans expressing the GFP fusion protein of interest. Prepare agarose pads on microscope slides. Paralyze worms using 2 mM levamisole in M9 buffer and transfer them to the pad.
- Microscopy Setup: Use an upright fluorescence microscope equipped with a color camera. Install the triple-band filter cube, which consists of:
- Excitation filter: ET485/10x (center wavelength 485 nm, bandwidth 10 nm).
- Dichroic beamsplitter: A triple-band mirror (e.g., Chroma 69000bs).
- Emission filter: A triple-band filter (e.g., Chroma 69000m) that allows light at 520/20 nm and 595/40 nm to pass but blocks the intermediate wavelengths.
- Image Acquisition: Capture images. With this setup, the true GFP signal will appear in green, while the autofluorescence will appear yellow, allowing for clear visual distinction and more accurate quantification.
Protocol 2: Assessing Bilirubin-Binding Protein Performance via Time-Gated Detection
This protocol leverages the long fluorescence lifetime of specialized dyes to separate their signal from short-lived autofluorescence [48].
Workflow:
- Probe Selection and Labeling: Conjugate your antibody or other targeting molecule with a long-lifetime fluorophore such as an ADOTA derivative (lifetime ~15 ns).
- Sample Staining: Label the tissue sample with the conjugated probe using standard immunofluorescence protocols.
- Time-Gated Image Acquisition: Use a microscope equipped with pulsed laser excitation and time-correlated single-photon counting (TCSPC) capability. Set a time gate to discard all photons detected within the first 10-20 nanoseconds after the excitation pulse.
- Analysis: The remaining signal originates predominantly from the long-lived probe, with over 96% of the autofluorescence removed. The signal-to-background ratio can be improved by fivefold or more [48].
The Scientist's Toolkit: Key Reagents and Materials
The following table details essential reagents and their functions for implementing the strategies discussed above.
Table 2: Essential Research Reagents for Autofluorescence Mitigation
| Reagent / Material | Primary Function | Key Consideration |
|---|---|---|
| DyLight 649 Conjugate [46] | A far-red emitting fluorophore that spectrally avoids common green autofluorescence. | A practical alternative to green fluorophores like DyLight 488. |
| Vector TrueVIEW Autofluorescence Quenching Kit [46] | Chemically quenches autofluorescence from non-lipofuscin sources in various tissue types. | Tissue-specific efficacy; particularly useful for problematic tissues like kidney, spleen, and pancreas. |
| Triple Band Filter Set (e.g., Chroma 69000) [47] | Optically separates GFP emission from tissue autofluorescence during image acquisition. | Requires a color camera for effective visualization of the separated colors. |
| ADOTA-NHS Fluorophore [48] | A long-lifetime (~15 ns) dye for time-gated detection to suppress short-lived autofluorescence. | Requires instrumentation capable of pulsed excitation and time-gated detection (e.g., TCSPC). |
| Sudan Black B [46] [50] | A chemical quenching agent used to reduce tissue autofluorescence. | Typically dissolved in 70% ethanol for application. |
| Sodium Borohydride [46] | A reducing agent used to diminish aldehyde-induced autofluorescence from fixation. | Used diluted in a physiological buffer like PBS or TBS. |
| Ethanol/Methanol Fixative [46] | An alternative to aldehyde-based fixatives that generates less autofluorescence. | May not be suitable for all antigens or experimental setups. |
Mitigating autofluorescence requires a strategic approach tailored to the specific experimental system. While GFP remains a powerful tool, its susceptibility to interference from common autofluorescence can be a major limitation. Bilirubin-binding proteins like UnaG offer advantages such as oxygen-independent maturation and high quantum efficiency, but they introduce a dependency on bilirubin availability.
The most robust solutions often combine multiple strategies. Selecting red-shifted fluorophores, optimizing sample preparation to minimize background at the source, and employing advanced optical or computational techniques for signal separation provide a comprehensive defense against autofluorescence. By understanding the strengths and limitations of different fluorescent proteins and the full arsenal of available tools, researchers can design experiments that deliver the clean, reliable signals necessary for groundbreaking discoveries in drug development and biomedical research.
Fluorescent proteins (FPs) have revolutionized molecular and cellular biology by enabling real-time observation of protein localization, dynamics, and interactions within living cells. Since the discovery and cloning of green fluorescent protein (GFP) from Aequorea victoria, these versatile tools have become indispensable for researchers studying gene expression, protein stability, and cellular architecture [29] [15]. The development of genetically encoded fluorescent tags allows scientists to monitor biological processes in live cells without the need for fixation and staining, providing unprecedented insight into dynamic cellular events.
The optimization of fusion protein design represents a critical challenge in biomedical research, particularly for drug development professionals requiring precise localization and unimpaired function of tagged proteins. While GFP and its variants have dominated the field for decades, newer alternatives like bilirubin-binding fluorescent proteins (BRFPs) such as UnaG offer unique advantages for specialized applications. This guide provides a comprehensive comparison of these systems, presenting experimental data and methodologies to inform selection criteria for specific research needs. The choice between different fluorescent protein systems significantly impacts experimental outcomes, particularly in studies involving anaerobic environments, quantitative imaging, or precise subcellular localization.
Fundamental Properties: GFP vs. Bilirubin-Binding UnaG
The selection of an appropriate fluorescent protein requires careful consideration of fundamental photophysical and biochemical properties. The following table summarizes key characteristics of GFP compared to the bilirubin-binding protein UnaG:
Table 1: Fundamental Properties of GFP and UnaG Fluorescent Proteins
| Property | GFP and Variants | UnaG (Bilirubin-Binding) |
|---|---|---|
| Fluorophore Formation | Autonomous, oxygen-dependent maturation [5] | Instantaneous, oxygen-independent upon bilirubin binding [5] [21] |
| Source | Aequorea victoria (jellyfish) and coral reefs [5] [6] | Anguilla japonica (Japanese eel) and other eels [5] [7] [6] |
| Molecular Structure | β-barrel with internal cyclized tripeptide [6] | β-barrel structure, member of fatty-acid-binding protein family [5] [7] |
| Molecular Weight | ~27 kDa (GFP) | 15.6 kDa [7] [6] |
| Ligand Requirement | None | Requires bilirubin (BR) [5] [7] |
| Quantum Efficiency | ~50% (UnaG equivalent to enhanced GFP) [5] | ~50% (highly efficient) [5] |
| Key Advantage | Well-established, no external cofactor | Function in anaerobic conditions [21] |
UnaG represents a distinct class of fluorescent proteins discovered in the muscle tissue of the Japanese eel, Anguilla japonica [7]. Unlike GFP, UnaG is a ligand-induced fluorescent protein (LIFP) that requires binding with bilirubin (BR) to emit fluorescence. The UnaG-bilirubin complex (holoUnaG) exhibits high quantum efficiency comparable to enhanced GFP but with the significant advantage of instantaneous fluorophore formation that does not depend on molecular oxygen [5]. This property makes UnaG particularly valuable for studying anaerobic biological systems where conventional FPs fail to fluoresce.
Performance Comparison: Experimental Data and Quantitative Analysis
Direct comparison of experimental performance metrics provides valuable insights for selecting appropriate fluorescent tags. The following table summarizes key performance characteristics based on published experimental data:
Table 2: Experimental Performance Comparison of Fluorescent Protein Systems
| Performance Metric | GFP and Variants | UnaG | Experimental Context |
|---|---|---|---|
| Maturation Time | Several hours (oxygen-dependent) [5] | Instantaneous (upon BR binding) [5] | Live-cell imaging [5] |
| Anaerobic Function | Limited or non-functional [21] | Fully functional [21] | Imaging of obligate anaerobes [21] |
| Brightness Ratio Between States | N/A | 1:3.9 (holoUnaG1:holoUnaG2) [5] | Photon counting analysis [5] |
| Oligomeric State in Solution | Variable (some form strong tetramers) [5] | Monomeric (both apo and holo states) [5] | Analytical ultracentrifugation [5] |
| Two-State Equilibrium Population | N/A | 6:4 (holoUnaG1:holoUnaG2) [5] | Spectroscopic analysis [5] |
| Detection Method Flexibility | Standard fluorescence detection [15] | Specialized bilirubin addition required [5] | In-gel fluorescence and live-cell imaging [5] [15] |
UnaG exhibits complex photophysical behavior with two distinct fluorescence states—holoUnaG1 and holoUnaG2—that exist in equilibrium with a brightness ratio of 1:3.9 and population ratio of 6:4 under experimental conditions [5]. This two-state system presents both challenges and opportunities for experimental design, potentially enabling more sophisticated biosensor applications. From a practical standpoint, UnaG maintains monomeric status in aqueous solution even at high salt concentrations (300 mM NaCl), reducing the risk of artifactual oligomerization that can impair the function of tagged proteins [5]. This characteristic represents a significant advantage over FPs like DsRed, which form obligate tetramers that can interfere with normal protein function [5].
Unique Mechanism: The Two-State Fluorescence of UnaG
UnaG exhibits a sophisticated fluorescence mechanism that distinguishes it from conventional fluorescent proteins. The protein exists in two distinct states—holoUnaG1 and holoUnaG2—that interconvert through a reversible intramolecular reaction [5]. The initial complex formed after bilirubin binding (holoUnaG1) transitions to a brighter state (holoUnaG2), with the two states reaching a specific equilibrium in solution. Spectroscopic analyses indicate that this transition is not associated with chemical changes to bilirubin but rather with alterations in the environmental conditions surrounding the fluorophore [5].
Diagram 1: UnaG Two-State Fluorescence Mechanism
The molecular brightness ratio between these states is approximately 1:3.9 (holoUnaG1:holoUnaG2), with an equilibrium population ratio of 6:4 as determined by photon number counting analysis [5]. This complex photophysical behavior enables potential engineering of UnaG variants with optimized brightness by stabilizing the brighter holoUnaG2 state. The diagram above illustrates the transition pathway between these fluorescent states, highlighting the reversible nature of the intramolecular reaction that modulates fluorescence intensity.
Experimental Protocols: Methodology for Fluorescent Protein Analysis
Protocol 1: UnaG-Bilirubin Binding and Fluorescence Analysis
Sample Preparation:
- Clone UnaG coding region into expression vector (e.g., pColdI with 6×His tag) [5]
- Express protein in E. coli BL21 cells with 0.5 mM IPTG induction at 16°C for 18 hours [5]
- Purify using HisTrap HP column with imidazole elution (20-500 mM gradient) [5]
- Prepare bilirubin stock solution (1 mM in DMSO, stored at -20°C) [5]
Fluorescence Assay:
- Add bilirubin to purified UnaG protein in potassium phosphate buffer (pH 7.5) [5]
- Monitor fluorescence time course using spectrophotometer (excitation 496 nm, emission 527 nm) [5]
- Record fluorescence intensity changes over time to observe holoUnaG1 to holoUnaG2 transition [5]
- Analyze data using photon counting to determine brightness ratio and equilibrium constants [5]
Key Considerations:
- Maintain bilirubin in dark conditions to prevent degradation
- Use fresh bilirubin solutions for consistent results
- Account for two-state system in quantitative measurements
Protocol 2: Subcellular Localization and Function Validation
Fusion Protein Design:
- Clone gene of interest in-frame with fluorescent protein at N- or C-terminus
- Consider terminal flexibility with linker sequences (e.g., GGGGS repeats)
- Select appropriate subcellular targeting sequences if needed
Localization Verification:
- Transfert fusion construct into appropriate cell line
- Image using confocal microscopy with appropriate filter sets
- Compare localization pattern with untagged protein or known markers
- Validate function through relevant biological assays
Anaerobic Imaging (for UnaG):
- Culture anaerobic bacteria in appropriate sealed chambers [21]
- Add bilirubin (for UnaG) or biliverdin (for IFP2.0) to growth medium [21]
- Image using standard fluorescence microscopy without oxygen requirement [21]
Research Reagent Solutions: Essential Materials for Experimental Success
Table 3: Key Research Reagents for Fluorescent Protein Studies
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Expression Vectors | pColdI, pGEX, pTolT expression vectors [5] [7] | Recombinant protein expression | pColdI suitable for 6×His-UnaG expression [5] |
| Chromatography Media | HisTrap HP column [5] | Protein purification | Efficient purification of histidine-tagged proteins |
| Fluorescent Protein Tags | GFP variants, UnaG, IFP2.0 [5] [21] | Protein tagging and visualization | UnaG ideal for anaerobic studies [21] |
| Ligands/Cofactors | Bilirubin, biliverdin [5] [21] | Activate bilin-binding FPs | Bilirubin for UnaG; biliverdin for IFP2.0 [21] |
| Inducers | IPTG, arabinose [5] | Induce protein expression | Concentration optimization required |
| Spectroscopy Equipment | RF-5300PC spectrophotometer, F-7100 fluorescence spectrometer [5] [7] | Fluorescence measurement | Key for quantification and characterization |
Application Workflows: Experimental Design and Implementation
The selection between GFP and bilirubin-binding proteins significantly impacts experimental design, particularly for specialized applications. The following diagram illustrates key decision points in selecting and implementing these fluorescent tools:
Diagram 2: Fluorescent Protein Selection Workflow
For anaerobic applications, UnaG provides unique capabilities as demonstrated in studies of prevalent gut bacterium Bacteroides thetaiotaomicron, where it enabled fluorescence labeling in oxygen-free environments [21]. This capability facilitates investigation of previously inaccessible anaerobic polymicrobial communities, such as those found in the human gut microbiome. Furthermore, UnaG can be utilized in multi-species, multicolor imaging approaches when combined with other bilin-binding proteins like IFP2.0 [21].
The choice between GFP and bilirubin-binding fluorescent proteins represents a critical strategic decision in experimental design. GFP variants remain excellent choices for standard aerobic applications with their well-characterized properties and extensive validation history. However, UnaG and related bilirubin-binding proteins offer distinct advantages for specialized applications, particularly in anaerobic environments, with additional benefits including instantaneous activation, monomeric structure, and unique two-state fluorescence properties.
Future directions in fluorescent protein development will likely focus on engineering improved UnaG variants with modulated binding affinity and fluorescence properties. Recent research has already demonstrated successful mutagenesis of UnaG residues (R112M, R132M, and R112&132M) to alter bilirubin-binding characteristics and thermal stability [7]. These advances, combined with emerging applications in live-cell imaging of anaerobic systems, position bilirubin-binding proteins as valuable additions to the molecular toolkit for drug development professionals and research scientists seeking to optimize fusion protein design for proper localization and function.
Head-to-Head Comparison: Validating Performance for Informed Choice
Fluorescent proteins (FPs) have revolutionized molecular and cellular biology by enabling real-time visualization of dynamic processes within living systems. Initially dominated by the green fluorescent protein (GFP) and its engineered variants from Aequorea victoria, the FP toolbox has expanded to include diverse protein families with distinct biochemical and photophysical properties [29]. Among these, bilirubin-binding fluorescent proteins (BBFPs), such as UnaG, represent a significant advancement with unique characteristics that address specific experimental limitations of conventional GFPs [5] [21]. This guide provides a direct, data-driven comparison of the performance metrics—specifically brightness, maturation time, and photostability—across leading GFP variants and emerging bilirubin-binding alternatives, offering researchers an evidence-based framework for selecting optimal probes for their experimental conditions.
The fundamental distinction between these FP classes lies in their chromophore requirements. Traditional GFP-like proteins form their chromophore autocatalytically from internal amino acids in a process that requires molecular oxygen, which can limit their use in anaerobic environments [21]. In contrast, BBFPs like UnaG are ligand-induced fluorescent proteins that bind the exogenous chromophore bilirubin (a product of heme catabolism) to fluoresce, a process that is instantaneous and oxygen-independent [5] [7]. This intrinsic difference underpins many of the performance trade-offs explored in this comparison and opens unique application spaces for each FP type.
Performance Matrix: Quantitative Comparison of Key Metrics
The following tables synthesize experimental data from systematic benchmarking studies to facilitate direct comparison of critical performance parameters across prominent fluorescent proteins.
Table 1: Maturation Kinetics of Common Fluorescent Proteins in E. coli [51]
| Fluorescent Protein | Class | 37°C t50 (min) | 37°C t90 (min) | 32°C t50 (min) | 32°C t90 (min) |
|---|---|---|---|---|---|
| mGFPmut3 | Green (avGFP-derived) | 4.1 ± 0.3 | 15.8 ± 3.1 | 4.5 ± 0.3 | 16.6 ± 2.9 |
| mVenus NB | Yellow-Green | 4.1 ± 0.3 | 18.4 ± 6.8 | 4.7 ± 0.4 | 18.0 ± 3.2 |
| sfGFP | Green (avGFP-derived) | 13.6 ± 0.9 | 39.1 ± 4.7 | 19.4 ± 1.3 | 56.7 ± 6.0 |
| mEGFP | Green (avGFP-derived) | 14.5 ± 1.0 | 42.4 ± 4.4 | 22.3 ± 1.5 | 62.8 ± 6.6 |
| mNeonGreen | Yellow-Green | 10.9 ± 0.8 | 36.8 ± 9.2 | 13.3 ± 0.9 | 37.7 ± 4.0 |
| mClover3 | Yellow-Green | 43.5 ± 2.9 | 112.4 ± 9.6 | 63.5 ± 3.6 | 176.5 ± 19.8 |
| mStayGold | Green (Engineered) | N/A | N/A | N/A | N/A |
| UnaG | Bilirubin-Binding | Instantaneous* | Instantaneous* | Instantaneous* | Instantaneous* |
Table 2: Brightness and Photostability Comparison in Live-Cell Imaging [16] [52]
| Fluorescent Protein | Class | Excitation Peak (nm) | Emission Peak (nm) | Relative Brightness (vs eGFP) | Relative Photostability |
|---|---|---|---|---|---|
| eGFP | Green (avGFP-derived) | 488 | 507 | 1.0 | Baseline |
| mNeonGreen | Yellow-Green | 506 | 517 | ~2-3x eGFP [52] | Lower than eGFP [52] |
| sfGFP | Green (avGFP-derived) | 485 | 510 | Similar to eGFP [16] | Similar to EGFP [16] |
| YuzuFP (sfGFP-H148S) | Green (avGFP-derived) | 485 | 510 | 1.5x sfGFP [16] | ~3x sfGFP [16] |
| mStayGold | Green (Engineered) | 491 | 511 | Highest among tested FPs [52] | Highest among tested FPs [52] |
| UnaG (holoUnaG2) | Bilirubin-Binding | 498 | 527 | QE ~50% (similar to eGFP) [5] | N/A |
Key Insights from Performance Data:
- Maturation Speed: Fastest-maturing GFPs (e.g., mGFPmut3, mVenus NB) achieve 50% maturity in ~4 minutes at 37°C, whereas slower variants (e.g., mClover3) require >40 minutes [51]. UnaG exhibits instantaneous fluorophore formation upon bilirubin binding [5].
- Temperature Dependence: Maturation kinetics for all GFPs are temperature-sensitive, proceeding approximately 1.5-2 times slower at 32°C versus 37°C [51].
- Brightness & Stability: Newer engineered GFPs like mStayGold and YuzuFP demonstrate significant improvements in both brightness and photostability compared to traditional eGFP and sfGFP [16] [52]. mNeonGreen, while bright, shows notably faster photobleaching [52].
Experimental Protocols for Key Performance Assays
Maturation Kinetics Measurement via Translation Arrest
Principle: This method quantifies maturation time by arresting new protein synthesis and monitoring the subsequent increase in fluorescence as pre-existing immature proteins complete their maturation process [51].
Detailed Protocol:
- Cell Preparation & Imaging: Grow E. coli expressing the target FP exponentially in a microfluidic device or agarose-based single-cell chemostat, allowing continuous observation of hundreds of cells over multiple generations.
- Translation Inhibition: Rapidly introduce chloramphenicol into the cell environment via precise microfluidic flow. Final concentration should completely inhibit bacterial translation (e.g., 100-200 µg/mL).
- Fluorescence Monitoring: Immediately after chloramphenicol addition, acquire time-lapse fluorescence images at short intervals (e.g., every 2-5 minutes) using minimal exposure time to minimize photobleaching.
- Data Analysis: For each cell, normalize the fluorescence intensity to the value at the moment of translation arrest. Plot the normalized fluorescence increase over time. Fit the resulting curve to extract the half-time (t50) and 90%-maturation time (t90), which represent the time required for 50% or 90% of the immature proteins to become fluorescent, respectively [51].
Technical Considerations:
- The observed maturation kinetics can be mono-exponential, bi-exponential, or exhibit more complex patterns, suggesting multiple rate-limiting steps in the chromophore formation pathway [51].
- This assay should be performed at multiple controlled temperatures (e.g., 32°C and 37°C) as maturation is highly temperature-dependent.
Photostability Assessment Under Constant Illumination
Principle: This test measures the resistance of a fluorescent protein to photobleaching by quantifying the decay of its fluorescence signal under continuous, high-intensity illumination.
Detailed Protocol:
- Sample Preparation: Express FPs in a standardized biological system (e.g., C. elegans head muscle, cultured mammalian cells) under isogenic conditions to ensure comparable expression levels and cellular environments.
- Pre-bleach Imaging: Capture a baseline fluorescence image using low-intensity light to avoid premature bleaching.
- Constant Illumination: Expose a defined region of interest (ROI) to high-power laser light (e.g., 80% laser power) for a set number of repeated cycles or continuous duration.
- Signal Quantification: After each bleaching cycle, capture a post-bleach image with the same low-intensity settings as the pre-bleach image.
- Data Analysis: Measure the mean fluorescence intensity within the ROI for each time point. Normalize to the pre-bleach intensity and plot the normalized fluorescence against time or cumulative light dose. The decay rate (e.g., time to 50% intensity loss) serves as the photostability metric [52].
Bilirubin Binding and Fluorescence Characterization for UnaG
Principle: This protocol characterizes the ligand-induced fluorescence of UnaG by titrating bilirubin and monitoring complex formation and the transition between its two distinct fluorescent states [5] [7].
Detailed Protocol:
- Protein Purification: Express and purify apoUnaG (the ligand-free, non-fluorescent form) in buffer. The 6xHis-tagged protein can be purified via Ni-NTA affinity chromatography.
- Ligand Titration: Prepare a concentrated stock of bilirubin in DMSO. Add increasing concentrations of bilirubin to fixed amounts of apoUnaG in solution.
- Spectroscopic Monitoring: After each bilirubin addition, record:
- Absorbance Spectrum: To monitor complex formation.
- Fluorescence Emission Spectrum: To quantify the development of green fluorescence (Em ~527 nm upon Ex ~498 nm).
- Kinetic Analysis: For a single bilirubin addition, monitor the fluorescence intensity over time to detect the reversible transition from the initially formed holoUnaG1 state to the brighter holoUnaG2 state. The molecular brightness ratio of these states is approximately 1:3.9, with an equilibrium population ratio of about 6:4 [5].
- Affinity Calculation: Plot the fluorescence intensity at maximum as a function of bilirubin concentration. Fit the binding isotherm to determine the dissociation constant (Kd).
Structural and Functional Relationships
The performance differences between fluorescent protein classes are rooted in their distinct structural characteristics and chromophore chemistries. The following diagram illustrates the key functional differences between GFP-like proteins and bilirubin-binding proteins like UnaG.
The Aequorea victoria GFP scaffold forms an 11-stranded β-barrel structure with an internal α-helix containing the chromophore-forming tripeptide. Chromophore maturation involves a multi-step autocatalytic process of cyclization, dehydration, and oxidation, requiring molecular oxygen and time, which limits its use in anaerobic conditions and for monitoring rapid gene expression dynamics [51] [9]. Key residues like H148 (mutated to serine in the brighter YuzuFP variant) interact with the chromophore and a conserved water molecule, significantly influencing brightness and photostability [16].
In contrast, UnaG belongs to the fatty-acid-binding protein family and also adopts a β-barrel structure. However, it remains non-fluorescent in its apo state until it binds bilirubin, a linear tetrapyrrole, in an oxygen-independent process [5] [7]. Upon binding, the UnaG-BR complex exists in two distinct fluorescent states (holoUnaG1 and holoUnaG2) that interconvert reversibly, with holoUnaG2 being nearly four times brighter [5]. This unique mechanism enables immediate fluorescence activation and operation in anaerobic environments, making it ideal for imaging gut microbiome bacteria and other obligate anaerobes [21].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Fluorescent Protein Research
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Chloramphenicol | Translation inhibitor for maturation kinetics assays. | Used in translation arrest assays to measure FP maturation half-times (t50, t90) [51]. |
| Bilirubin (BR) | Exogenous chromophore ligand for UnaG activation. | Titrated into apoUnaG to induce fluorescence; enables imaging in anaerobic conditions [5] [21]. |
| Biliverdin (BV) | Exogenous chromophore for near-infrared FPs (e.g., IFP2.0). | Used with BBFPs for multi-color anaerobic imaging; expands spectral palette [21]. |
| Microfluidic Devices / Single-Cell Chemostats | Precise cell culture environment for time-lapse imaging. | Enables high-precision maturation kinetics measurements in growing cells under controlled conditions [51]. |
| Site-Directed Mutagenesis Kits | Protein engineering tool for FP optimization. | Used to create point mutations (e.g., H148S in YuzuFP) to improve brightness and photostability [16] [7]. |
| Affinity Chromatography Resins | Protein purification (e.g., HisTrap for 6xHis-tagged FPs). | Essential for purifying recombinant FPs like UnaG for in vitro spectroscopic characterization [5]. |
Application-Based Selection Guidelines
Choosing the optimal fluorescent protein requires balancing multiple performance characteristics against specific experimental needs. The following decision pathway provides a systematic approach for researchers to select the most suitable FP based on key application requirements.
Key Selection Criteria:
- For Anaerobic or Hypoxic Studies: Bilirubin-binding proteins like UnaG and near-infrared BBFPs like IFP2.0 are unequivocally superior due to their oxygen-independent fluorescence. They enable live-cell imaging of obligate anaerobes such as prevalent gut microbiome species (Bacteroides thetaiotaomicron) and investigations of hypoxic tissue environments [21].
- For Rapid Dynamics and High Temporal Resolution: Fast-maturing GFPs such as mGFPmut3 and mVenus NB (t50 ~4 minutes) are essential for accurately monitoring rapid gene expression changes and protein turnover, as slow maturation (e.g., mClover3 with t50 >40 minutes) introduces significant temporal lag and effectively reduces signal in growing cells [51].
- For Maximum Brightness and Photostability: The newly engineered mStayGold represents the current state-of-the-art, demonstrating superior brightness and exceptional resistance to photobleaching in direct comparisons with eGFP, mNeonGreen, and GFPnovo2 in C. elegans models [52]. For applications where mStayGold is incompatible (e.g., with nanobody-based systems), YuzuFP offers a compelling alternative with 1.5x the brightness and 3x the photobleaching resistance of its parent sfGFP [16].
- For General-Purpose Use: Well-characterized workhorses like sfGFP and mEGFP remain excellent choices for many standard applications, offering a reliable balance of brightness, maturation speed, and compatibility with existing laboratory protocols and detection systems.
The ongoing diversification of the fluorescent protein toolkit, from engineered GFP variants to structurally distinct bilirubin-binding proteins, provides researchers with an expanding array of optimized tools for live-cell imaging. Performance-critical applications now demand careful matching of FP characteristics to experimental conditions, whether prioritizing speed (maturation kinetics), signal intensity (brightness), duration (photostability), or environmental compatibility (aerobic/anaerobic). The quantitative data and decision framework presented here offer a evidence-based approach for selecting the optimal probe, acknowledging that the ideal fluorescent protein is ultimately defined by the specific biological question and experimental system under investigation. As protein engineering continues to advance, further breakthroughs in FP performance will undoubtedly emerge, but the fundamental trade-offs between different protein classes will likely continue to inform their appropriate application spaces.
In the toolkit of modern bioscience, fluorescent proteins have revolutionized our ability to visualize cellular processes in real-time. While Green Fluorescent Protein (GFP) and its derivatives from Aequorea victoria represent one well-established paradigm, a distinct class of ligand-induced fluorescent proteins has emerged, offering unique properties and challenges [29]. Among these, UnaG—a fatty acid-binding protein derived from the Japanese eel (Anguilla japonica)—exemplifies a fundamentally different mechanism: it fluoresces not through an intrinsic chromophore formed by autocatalysis, but only upon binding the endogenous metabolite bilirubin [7] [5]. This critical difference creates a research landscape where the availability of the bilirubin cofactor becomes a primary experimental consideration, directly influencing signal intensity and detection sensitivity. This guide objectively compares the performance of bilirubin-dependent UnaG against traditional GFP-like proteins, providing researchers with the experimental data and protocols needed to inform their selection for specific applications.
Molecular Mechanisms: Intrinsic Chromophore vs. Inducible Fluorescence
The fundamental distinction between GFP and UnaG lies in their fluorescence mechanisms, which dictates their experimental requirements and potential applications.
GFP and Its Derivatives: Intrinsic Chromophore Formation
Proteins of the GFP family function as genetically encoded labels that form their chromophore through an autocatalytic, post-translational modification of their own amino acids. This process typically requires molecular oxygen and can take several hours to mature fully [5]. Once formed, the chromophore is covalently bound within the β-barrel structure of the protein, resulting in stable, constitutive fluorescence that is largely independent of external cellular factors beyond oxygen [45].
UnaG: Bilirubin-Induced Fluorescence
UnaG belongs to the family of ligand-induced fluorescent proteins (LIFPs) and remains non-fluorescent in its apo state (apoUnaG). Its fluorescence is activated specifically upon non-covalent binding to unconjugated bilirubin (UC-BR), forming the holoUnaG complex [7] [11]. The resulting fluorescence exhibits a high quantum efficiency of approximately 50%, comparable to enhanced GFP [5]. A unique characteristic of the holoUnaG complex is its existence in two distinct fluorescence states—holoUnaG1 and holoUnaG2—which undergo a reversible intramolecular reaction. The brighter holoUnaG2 state has a molecular brightness approximately 3.9 times greater than the initial holoUnaG1 state [5]. The following diagram illustrates this activation pathway and the associated cellular processes that influence bilirubin availability.
Diagram Title: UnaG Activation and Bilirubin Metabolism Pathway
Performance Comparison: Experimental Data and Applications
The dependence of UnaG on bilirubin shifts the experimental considerations from protein expression alone to a system that includes cofactor availability. The table below summarizes key performance characteristics based on experimental data.
| Feature | GFP/EGFP | UnaG |
|---|---|---|
| Fluorophore Type | Intrinsic, autocatalytic | Bilirubin (BR), non-covalent |
| Maturation | Oxygen-dependent, requires hours | Instantaneous upon BR binding |
| Excitation/Emission Maxima | ~488/507 nm [45] | ~498-504/527-542 nm [7] [5] |
| Quantum Efficiency | High (e.g., EGFP ~50%) [45] | High (~50%, equivalent to EGFP) [5] |
| Signal Dependency | Expression level, O₂ | Expression level, BR concentration |
| Key Advantage | Stable, constitutive signal | Oxygen-independent; specific BR sensing |
| Primary Limitation | Slow maturation; oxygen requirement | Signal dependent on endogenous BR or supplementation |
Functional Implications of Performance Data
The data in the table highlights critical operational differences. UnaG's instantaneous activation is a significant advantage in time-sensitive experiments or under hypoxic conditions where GFP maturation may be impaired [5]. Furthermore, the brightness of UnaG is sufficient for sensitive detection, as demonstrated in flow cytometry panels where it performs comparably to other bright fluorescent proteins [45]. However, the direct correlation between UnaG fluorescence and bilirubin concentration transforms it from a simple label into a quantitative biosensor [11] [53]. This property has been leveraged in clinical assays for directly measuring unconjugated bilirubin in newborn serum, demonstrating high correlation with conventional methods and robustness against interfering factors like hemoglobin or lipid emulsion [11].
Essential Research Reagents and Experimental Toolkit
Successful experimentation with UnaG requires reagents and controls that account for bilirubin metabolism and detection. The following table outlines a core toolkit.
| Reagent / Material | Function / Explanation | Experimental Context |
|---|---|---|
| UnaG Protein (Purified) | Core fluorescent reporter; binds bilirubin with high specificity. | Used in bilirubin quantification assays in lysates or serum [11] [53]. |
| Bilirubin (Unconjugated) | Essential fluorogenic ligand; required for activating UnaG fluorescence. | Used to generate standard curves for quantitative measurements [53]. |
| Biliverdin | Immediate precursor to bilirubin in the heme catabolism pathway. | Substrate for measuring BVR enzyme activity in functional assays [53]. |
| β-NADPH | Cofactor for Biliverdin Reductase A (BVRA). | Essential for in vitro BVR activity assays that generate bilirubin [53]. |
| BVR Assay Buffer | Optimizes enzymatic activity for bilirubin production. | Provides appropriate pH (Tris-based, pH 8.7) for BVRA function [53]. |
| Putative Inhibitors | Chemical tools to probe bilirubin production pathway. | Closantel and Ebselen used to inhibit BVRA and validate assay specificity [53]. |
Key Experimental Protocols for Bilirubin-Dependent Assays
Detailed methodologies are critical for generating reproducible and reliable data. The protocols below are adapted from foundational papers.
Protocol: Measuring Intracellular Bilirubin and BVR Activity with UnaG
This fluorescence-based assay is significantly more sensitive than traditional absorbance methods for measuring biliverdin reductase (BVR) activity and intracellular bilirubin [53].
Protein Lysate Preparation:
- Harvest cells or tissues and lyse using RIPA buffer supplemented with protease and phosphatase inhibitors.
- Centrifuge the homogenate at 16,000 × g to clarify and determine the protein concentration of the supernatant.
Reaction Setup:
- Samples: Combine cell/tissue lysate (300-400 µg of protein) with BVR assay buffer for a total volume of 250 µL.
- Bilirubin Standards: Prepare a separate set of tubes with 250 µL of fresh bilirubin standards in PBS (e.g., 0, 20, 200, 2000, 20,000 nM).
- Master Mix: Prepare a 2X BVR assay master mix containing:
- Biliverdin (60 µM final in 2X mix)
- β-NADPH (1.6 mg/mL final in 2X mix)
- Purified UnaG protein (70 µg/mL final in 2X mix)
- BVR assay buffer to volume.
- Add 250 µL of the 2X master mix to each sample and standard tube.
Incubation and Measurement:
- Incubate the reaction mixture to allow the enzymatic conversion of biliverdin to bilirubin by BVR in the samples. The bilirubin produced immediately binds to UnaG, generating fluorescence.
- Transfer the solutions to a microplate reader and measure the fluorescence (Ex ~500 nm / Em ~527 nm).
- Calculate bilirubin production in samples by interpolating from the bilirubin standard curve.
Protocol: Direct Detection of Unconjugated Bilirubin in Serum
This protocol outlines a robust method for using UnaG to directly measure unconjugated bilirubin in biological fluids like serum, which is crucial for clinical research applications [11].
- Sample Collection: Collect serum or plasma samples using standard clinical procedures.
- UnaG Reaction: Mix a small volume of serum (e.g., 1-5 µL) with a solution of purified UnaG protein in a suitable buffer.
- Kinetic Measurement: Monitor the fluorescence intensity (Ex ~500 nm / Em ~527 nm) over time. The signal typically stabilizes around 10 minutes after mixing.
- Quantification: Determine the unconjugated bilirubin concentration in the sample by comparing the fluorescence signal to a standard curve generated with known concentrations of unconjugated bilirubin in the same matrix.
The choice between GFP and a bilirubin-binding protein like UnaG is not a matter of which is universally superior, but which is optimal for a specific research context. Researchers must weigh the cofactor considerations against their experimental needs.
- Select GFP-based proteins for experiments requiring a consistent, constitutive fluorescent tag whose signal is primarily a function of promoter activity and protein expression, and where oxygen availability is not a limiting factor.
- Choose UnaG in scenarios where its unique properties are decisive: for imaging under hypoxic conditions; when a rapid, oxygen-independent fluorescence turn-on is critical; or, most importantly, when the experimental goal is to directly sense, quantify, or leverage the dynamics of bilirubin itself, a molecule with growing significance in antioxidant defense and cell signaling [54] [53].
The development of UnaG mutants with altered bilirubin binding affinity and fluorescence properties promises to further expand this toolkit, allowing researchers to fine-tune the protein's characteristics for specific physiological detection conditions [7].
The selection of an appropriate fluorescent protein (FP) is a critical decision in the design of in vivo imaging experiments. While Green Fluorescent Protein (GFP) and its derivatives have been workhorses in live-cell imaging for decades, their performance in deep-tissue applications can be limited. The emergence of alternative FPs, such as bilirubin-binding proteins (e.g., UnaG), offers distinct advantages for specific experimental conditions, particularly those involving hypoxic environments or requiring greater tissue penetration. This guide provides an objective comparison of GFP-based proteins versus bilirubin-binding fluorescent proteins, presenting key experimental data to help researchers select the optimal probe for their in vivo imaging applications. Performance is evaluated based on spectral properties, brightness, environmental sensitivity, and practical performance in biological systems, with all quantitative data summarized in structured tables for direct comparison.
Comparative Performance Data of Fluorescent Proteins
Table 1: Key Characteristics of Green Fluorescent Proteins
| Protein Name | Class | Excitation/Emission (nm) | Maturation Requirement | Molecular Size (kDa) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| GFP/EGFP | GFP-like | ~488/~510 [55] | Oxygen-dependent [5] | ~25-27 [19] | Bright; well-characterized [55] | Limited use in hypoxia/anoxia [5] |
| mNeonGreen | GFP-like | ~506/~517 [55] | Oxygen-dependent | ~26 | High quantum yield in vitro [55] | Lower in vivo brightness than predicted [55] |
| miniGFP1/2 | Flavin-binding | 450/499 [19] | Oxygen-independent [19] | ~13 [19] | Small size; works in anaerobes [19] | Lower brightness vs. GFP-like FPs [19] |
Table 2: Key Characteristics of Bilirubin-Binding Proteins and Other Alternatives
| Protein Name | Class | Excitation/Emission (nm) | Maturation Requirement | Molecular Size (kDa) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| UnaG | Bilirubin-binding | Not specified in results | Oxygen-independent; requires bilirubin [5] | Monomer [5] | Instant fluorogen formation; high quantum efficiency (~50%) [5] | Requires exogenous bilirubin [5] |
| HoloUnaG1 | Bilirubin-bound state | Not specified in results | Instantaneous [5] | Monomer [5] | First formed state [5] | Lower brightness [5] |
| HoloUnaG2 | Bilirubin-bound state | Not specified in results | Forms from HoloUnaG1 [5] | Monomer [5] | Brighter state (1:3.9 ratio to HoloUnaG1) [5] | Equilibrium population ratio (6:4, HoloUnaG1/HoloUnaG2) [5] |
| Flavins (e.g., LOV-based) | Flavin-binding | Varies | Oxygen-independent [29] [19] | ~12-16 [19] | Small size; anaerobic imaging [19] | Limited brightness [19] |
Table 3: Quantitative In Vivo Performance Comparison in C. elegans Embryos
| Fluorescent Protein | Excitation (nm) | Relative In Vivo Brightness | Notes |
|---|---|---|---|
| GFP | 488 | High | Nearly twice as bright as mNeonGreen with 488nm excitation [55] |
| mNeonGreen | 488 | Low | Not as bright in vivo as predicted by in vitro data [55] |
| mNeonGreen | 514 | Low | About four times less bright than mYPet with 514nm excitation [55] |
| mYPet | 514 | High | Superior performance with 514nm excitation [55] |
| Background Autofluorescence | 405/442 | Lowest | Not recommended for live imaging due to phototoxicity [55] |
| Background Autofluorescence | 488 | Highest | Significant background noise [55] |
| Background Autofluorescence | 514 | Medium | Reduced background compared to 488nm [55] |
Experimental Protocols for Key Comparisons
Protocol: Assessing Fluorescent Protein Brightness and Photostability In Vivo
This protocol is adapted from methods used to quantitatively compare FPs in C. elegans embryos [55].
1. Sample Preparation:
- Generate transgenic strains expressing the FPs of interest as single-copy knock-ins at the same genomic locus to ensure consistent expression levels.
- For the C. elegans model, use CRISPR/Cas9-triggered homologous recombination to insert the transgene.
- Confirm knock-ins by observing the expected fluorescence localization pattern and verify single-copy insertion by PCR genotyping and sequencing.
2. Imaging Setup:
- Use a spinning-disk confocal microscope equipped with standard lasers (e.g., 488nm, 514nm) and an EM-CCD camera.
- For quantitative comparisons, mount staged embryos from different strains side-by-side on the same slide.
- Maintain identical imaging conditions (laser power, exposure time, camera gain, filter sets) for all samples.
3. Data Acquisition:
- Acquire images of the embryos under the same non-saturating conditions.
- To assess photostability, perform time-lapse imaging under continuous illumination, monitoring fluorescence decay over time.
4. Data Analysis:
- Quantify fluorescence intensity by measuring mean pixel values in a defined region of interest (e.g., embryo membrane).
- Normalize fluorescence intensities to the autofluorescence levels of wild-type embryos.
- For photostability, calculate the time for fluorescence intensity to decay to 50% of its initial value (t½).
Protocol: Testing Protein Performance under Hypoxic and Anaerobic Conditions
This protocol is based on experiments demonstrating the utility of flavin-binding FPs for imaging under low oxygen [19].
1. Cell Culture and Transfection:
- Culture mammalian cells (e.g., CHO cells) expressing the FPs of interest under standard conditions.
- For bacterial expression, transform E. coli with plasmids encoding the FPs.
2. Induction of Low-Oxygen Conditions:
- For mammalian cells, place culture plates in a hypoxia chamber with a gas mixture containing 1-5% O₂ for several hours to days.
- For anaerobic bacterial imaging, use an anaerobic chamber or seal cultures in an airtight system with oxygen scavengers.
3. Imaging and Analysis:
- Image live cells/bacteria under the respective conditions using a fluorescence microscope equipped with appropriate filter sets.
- Compare fluorescence intensity and signal-to-noise ratio to control samples maintained under normoxic conditions.
- For quantitative analysis, use flow cytometry to measure fluorescence intensity in a large cell population.
Protocol: Characterizing Ligand-Induced Fluorescent Proteins
This protocol outlines the procedure for studying bilirubin-binding proteins like UnaG, based on published methodologies [5].
1. Protein Purification:
- Express 6xHis-tagged UnaG in E. coli BL21 cells induced with 0.5 mM IPTG at 16°C for 18 hours.
- Purify the protein using nickel affinity chromatography (HisTrap HP column) with imidazole elution.
- Desalt the eluate into an appropriate buffer (e.g., 20 mM potassium phosphate, pH 7.5).
2. Ligand Binding Kinetics:
- Prepare a 1 mM bilirubin stock solution in DMSO.
- Mix UnaG with bilirubin at desired concentrations and rapidly measure fluorescence intensity over time.
- Monitor the transition between the two distinct fluorescence states (holoUnaG1 and holoUnaG2).
3. Oligomeric State Determination:
- Perform analytical ultracentrifugation sedimentation velocity (AUC-SV) and sedimentation equilibrium (AUC-SE) analysis.
- Use protein concentrations of 15 μM for AUC-SV and 3.8-9.4 μM for AUC-SE.
- Analyze data using SEDFIT and ORIGIN software to determine molecular mass and oligomeric state.
Visualizing Fluorescent Protein Workflows and Properties
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for In Vivo Fluorescence Imaging
| Reagent/Resource | Function/Application | Examples/Specifications |
|---|---|---|
| Fluorescent Protein Plasmids | Expression vectors for FP fusion proteins | Commercially available FP plasmids (e.g., Addgene); codon-optimized for target organisms |
| Bilirubin (BR) | Ligand for UnaG activation | 1 mM stock solution in DMSO; required for UnaG fluorescence [5] |
| Flavin Mononucleotide (FMN) | Endogenous chromophore for flavin-binding FPs | Naturally present in cytoplasm; enables oxygen-independent fluorescence [19] |
| CRISPR/Cas9 Systems | For precise genomic integration of FP tags | Enables single-copy, endogenous tagging for quantitative comparisons [55] |
| Hypoxia Chambers | Creating low-oxygen environments for testing | Controlled atmosphere (1-5% O₂) for hypoxic condition studies [19] |
| Anaerobic Chambers | Creating oxygen-free environments | Essential for testing FPs in obligate anaerobes [19] |
| Spectrum Viewer Tool | Predicting FP performance with specific filter sets | Excel-based tool for matching FP spectra with imaging hardware [55] |
| Anti-FP Nanobodies | Detection and manipulation of FP-tagged proteins | Used in immunoprecipitation, western blot, and protein manipulation [15] |
The choice between GFP-like proteins, bilirubin-binding proteins, and other specialized FPs depends heavily on the specific experimental requirements. GFP variants remain excellent choices for well-oxygenated systems where high brightness is paramount. In contrast, bilirubin-binding UnaG offers unique advantages for hypoxic conditions and instantaneous activation, while flavin-binding proteins like miniGFPs enable imaging in anaerobic environments and provide sensitivity to metal ions. The experimental data and protocols presented here provide a framework for evidence-based selection of fluorescent proteins, considering not just their intrinsic brightness but also their performance under biologically relevant conditions. As in vivo imaging technologies continue to advance, particularly with developments in near-infrared imaging [56], the optimal FP choice will increasingly depend on the match between protein properties and the specific physiological environment being studied.
For decades, the green fluorescent protein (GFP) and its variants have been indispensable tools for life science research, enabling dynamic visualization of cellular processes. However, the emergence of bilirubin-inducible fluorescent proteins like UnaG and GymFP has expanded the fluorescent toolkit, offering unique solutions where classical GFPs fall short. This guide provides an objective, data-driven comparison of these technologies to help you select the optimal reporter for your specific research goals, from live-cell imaging under hypoxia to quantitative reporting in biofilms.
Fundamental Mechanisms and Properties
The core distinction between these protein classes lies in their chromophore generation and cofactor requirements, which directly dictate their experimental applications.
GFP Family Proteins: Oxygen-Dependent Intrinsic Chromophores
Proteins of the GFP family serve as genetically encoded labels that have brought fluorescence molecular imaging into the dynamic world of living cells and organisms [29]. The GFP from Aequorea victoria possesses a chromophore that forms through an autocatalytic, oxygen-dependent maturation process [13]. This process requires molecular oxygen for a final dehydrogenation step, which is essential for the protein to become fluorescent [57]. While enhanced GFP (EGFP) and other mutants have improved characteristics, this fundamental oxygen requirement remains a limitation for anaerobic applications [13].
Bilirubin-Binding Proteins: Ligand-Induced Fluorescence
In contrast, bilirubin-binding fluorescent proteins (BBFPs) such as UnaG and the recently discovered GymFP belong to the fatty acid-binding protein family and are the first FPs derived from vertebrates [5] [6]. These proteins remain in a non-fluorescent apo-state until they specifically bind bilirubin (BR), a breakdown product of heme metabolism, forming a highly fluorescent complex [5] [6]. This ligand-induced fluorescence is instantaneous and oxygen-independent, as it does not involve a post-translational oxidation step [5] [21].
Table: Fundamental Characteristics of GFP vs. Bilirubin-Binding Fluorescent Proteins
| Characteristic | GFP Family | Bilirubin-Binding Proteins |
|---|---|---|
| Chromophore Source | Intrinsic formation from amino acids | Exogenous bilirubin ligand |
| Maturation Process | Autocatalytic, oxygen-dependent | Instantaneous upon bilirubin binding |
| Oxygen Requirement | Essential | None |
| Native Source | Jellyfish (Aequorea victoria) | Eels (Anguilla japonica, Gymnothorax zonipectis) |
| Protein Family | GFP-like β-barrel | Fatty Acid Binding Protein (FABP) |
| Key Structural Motif | Ser-Tyr-Gly cyclization | Gly-Pro-Pro (GPP) motif for bilirubin protection |
Quantitative Performance Comparison
Understanding the spectral and biophysical properties of these fluorescent markers is crucial for experimental design, particularly for microscopy and quantitative applications.
Spectral Characteristics and Brightness
The recently discovered GymFP from the moray eel Gymnothorax zonipectis exhibits excitation and emission maxima at 496 nm and 532 nm respectively upon bilirubin addition [6]. This places it firmly in the green spectrum, similar to many GFP variants. The UnaG-BR complex demonstrates high quantum efficiency of approximately 50%, comparable to enhanced GFP, one of the brightest GFP mutants [5].
Research has revealed that the holoUnaG complex exists in two distinct fluorescence states—holoUnaG1 and holoUnaG2—with a molecular brightness ratio of 1:3.9 and an equilibrium population ratio of 6:4 [5]. This dynamic equilibrium between states with different brightness levels adds complexity to quantitative measurements using UnaG.
Oligomeric State and Fusion Compatibility
A critical advantage of UnaG for protein tagging applications is its monomeric nature. Analytical ultracentrifugation studies have confirmed that both apoUnaG and holoUnaG exist as monomers in aqueous solution even under conditions of 300 mM NaCl [5]. This monomeric state prevents aberrant oligomerization that can disrupt the function or localization of fusion proteins, a significant advantage over early fluorescent proteins like DsRed, which formed strong tetramers that disturbed some functions of labeled proteins in cells [5].
Table: Photophysical and Biochemical Properties Comparison
| Property | EGFP | UnaG | GymFP |
|---|---|---|---|
| Excitation Maximum (nm) | 488 [45] | 498 [5] | 496 [6] |
| Emission Maximum (nm) | 507 [45] | 509 [5] | 532 [6] |
| Quantum Yield | 0.60 [13] | ~0.50 [5] | Not specified |
| Extinction Coefficient (M⁻¹cm⁻¹) | 55,000 [13] | Not specified | Not specified |
| Oligomeric State | Monomeric variants available [13] | Monomeric [5] | Not specified |
| Molecular Mass (kDa) | 27 [13] | Not specified | 15.6 [6] |
| Chromophore Maturation Time | Minutes to hours (oxygen-dependent) [57] | Instantaneous (bilirubin-dependent) [5] | Instantaneous (bilirubin-dependent) [6] |
Experimental Applications and Performance Data
The distinct biochemical properties of these fluorescent proteins translate directly into differentiated performance across experimental conditions.
Anaerobic and Hypoxic Environments
The oxygen-independent fluorescence of BBFPs provides a decisive advantage in anaerobic environments where GFP fails. Conventional fluorescent proteins like GFP are restricted to aerobic environments due to their oxygen-dependent chromophore maturation [21]. This limitation precludes fluorescence studies of anaerobic ecologies including polymicrobial communities in the human gut microbiome and in soil microbiomes [21].
BBFPs have been successfully implemented for live-cell imaging of obligate anaerobic bacteria, such as the prevalent gut bacterium Bacteroides thetaiotaomicron, under truly anaerobic conditions [21]. Furthermore, these BBFPs enable two-color imaging to differentiate cells expressing either UnaG or IFP2.0, allowing researchers to distinguish between different bacterial species in mixed-culture live-cell imaging of anaerobic communities [21].
Quantitative Reporting in Biofilms and High-Density Cultures
GFP-like proteins exhibit significant limitations for quantitative measurements in high-density cultures like biofilms. Studies demonstrate that both GFP and mCherry exhibit fluorescence kinetics that quickly become inconsistent with cell biomass development in biofilms [57]. The spatial-temporal distribution of GFP fluorescence does not linearly report on gene expression in the confined environment of adherent bacterial communities [57].
This limitation stems from oxygen shortage within developing biofilms. When biofilm grows and cell oxygen consumption increases, the level of oxygen decreases below the threshold required for effective GFP fluorescence maturation [57]. In contrast, the fluorescence of BBFPs remains linearly correlated with protein expression regardless of oxygen gradients, making them superior for quantitative studies in biofilms and other high-density cultures [57].
Bilirubin Sensing and Analytical Applications
UnaG has enabled novel analytical applications beyond cellular imaging. A versatile fluorometric method for nanoscale analysis of bilirubin has been developed based on its highly specific binding to the recombinant bifunctional protein HELP–UnaG (HUG) [58]. This assay is remarkably sensitive (LoQ = 1.1 nM), accurate (4.5% relative standard error), and robust, allowing analysis at pH 7.4–9.5, T = 25–37 °C, in various buffers, and in the presence of serum albumin or DMSO [58].
This HUG-based assay can determine bilirubin in human plasma with a relative standard error of 6.7% at values that correlate and agree with the standard diazo method, demonstrating its suitability for translational and precision medicine applications [58].
Experimental Protocols and Methodologies
Implementing UnaG for Anaerobic Bacterial Imaging
To implement BBFPs for oxygen-independent fluorescent labeling in anaerobic bacteria [21]:
Genetic Construction: Clone the UnaG gene into an appropriate expression vector for your target organism. For Bacteroides thetaiotaomicron, use a Bacteroides-compatible vector with an appropriate antibiotic resistance marker.
Transformation: Introduce the construct into your target anaerobic bacterium using species-specific transformation protocols.
Ligand Application: Add bilirubin (water-soluble preparations are available) to the growth medium at optimal concentrations. For anaerobic work, prepare bilirubin solutions in an anaerobic chamber to prevent oxidation.
Anaerobic Cultivation: Grow transformed bacteria under strict anaerobic conditions using standard anaerobic culturing techniques (anaerobic chambers, pre-reduced media).
Imaging: Image living cells using standard fluorescence microscopy equipment with FITC filter sets or equivalent. No oxygen is required for fluorescence development.
Bilirubin Quantification Assay Using HUG Fusion Protein
The HUG-based assay enables sensitive bilirubin measurement across diverse experimental conditions [58]:
Protein Preparation: Express and purify the HELP-UnaG (HUG) bifunctional fusion protein, which retains both UnaG's bilirubin-dependent fluorescence and HELP's thermo-responsive behavior.
Standard Solution Preparation:
- Prepare 5 mM bilirubin stock in DMSO (Solution A)
- Dilute to 10 μM in PBS with BSA (Solution B1, pre-calibrator)
- Further dilute to nM range in PBS with BSA (Solution C1, calibrators)
- Include BSA (4 g/L) in solutions to stabilize bilirubin
Quality Control:
- Verify stock concentration by diluting to 10 μM BR in PBS-BSA and analyzing by diazo reaction using molar extinction coefficient of 76,490 cm⁻¹·M⁻¹
- Alternatively, use direct UV-Vis spectroscopy with ε = 61,079 cm⁻¹·M⁻¹ in PBS-BSA at 465 nm
Assay Performance:
- Working range: 1.1 nM to micromolar concentrations
- pH tolerance: 7.4-9.5
- Temperature tolerance: 25-37°C
- Compatible with various buffers and up to 30% DMSO
Research Reagent Solutions
Table: Essential Materials for Implementing Fluorescent Protein Technologies
| Reagent/Material | Function/Application | Implementation Notes |
|---|---|---|
| UnaG Expression Vector | Heterologous expression of bilirubin-inducible fluorescent protein | Available from academic repositories; codon-optimize for target organism [5] |
| Bilirubin (BR) | Ligand for UnaG activation | Dissolve in DMSO at 1 mM; store at -20°C protected from light [5] |
| HELP-UnaG (HUG) Fusion | Bilirubin quantification assay | Thermo-responsive fusion protein for sensitive BR detection [58] |
| Anaerobic Chamber | Creating oxygen-free environment for anaerobic imaging | Essential for culturing and imaging strict anaerobes with BBFPs [21] |
| EGFP/mCherry Vectors | Conventional fluorescent protein controls | Include as oxygen-sensitive controls for comparative studies [57] |
| Bovine Serum Albumin (BSA) | Bilirubin stabilization in solution | Use at 4 g/L in PBS for standard solutions; prevents BR aggregation [58] |
| Ruthenium Complex Micelles | Oxygen sensing in biofilms | Use with fluorescence lifetime microscopy to map O₂ gradients [57] |
When selecting between GFP and bilirubin-binding fluorescent proteins, consider these critical factors:
Oxygen Environment: For anaerobic or hypoxic conditions, BBFPs are unequivocally superior [21] [57]. In well-oxygenated systems, both options are viable.
Quantitative Requirements: For precise quantification in high-density cultures, biofilms, or environments with potential oxygen gradients, BBFPs provide more reliable linear reporting [57].
Experimental Simplicity: GFP variants offer simpler implementation for standard aerobic applications without requiring exogenous ligand addition.
Specialized Applications: For bilirubin sensing, metabolic studies of heme catabolism, or anaerobic community imaging, BBFPs enable unique experimental approaches not possible with GFP [21] [58].
The expanding palette of genetic markers, including flavin-, bilirubin-, and biliverdin-binding fluorescent proteins, continues to enrich our toolbox for biological imaging [29]. By carefully matching the properties of these fluorescent reporters to your specific experimental system and research questions, you can optimize the reliability and significance of your findings across diverse biological contexts from aerobic tissue culture to anaerobic microbiomes.
Conclusion
GFP and bilirubin-binding fluorescent proteins are not merely interchangeable tools but specialized technologies for distinct research niches. GFP remains the versatile, oxygen-dependent workhorse for standard live-cell imaging, while bilirubin-binding proteins like UnaG offer a powerful alternative for anaerobic studies, deeper tissue imaging, and contexts where GFP's pH sensitivity or oxygen requirement is prohibitive. The future of fluorescent tagging lies in the continued engineering of both systems to enhance brightness, photostability, and chromatic diversity, as well as in their innovative application for monitoring advanced cell and gene therapies. By understanding their complementary strengths and limitations, researchers can strategically select the optimal probe to illuminate their specific biological questions, driving discovery in basic science and accelerating translational progress.
References
- 1. Simulating green fluorescent proteins: The potential of ... [https://arabjchem.org/simulating-green-fluorescent-proteins-the-potential-of-fluorescent-aptamers-and-peptides-for-biosensing-and-imaging/]
- 2. Beta-Barrel Scaffold of Fluorescent Proteins: Folding, Stability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3739439/]
- 3. Correlation between fluorescence and structure in the ... [https://www.sciencedirect.com/science/article/pii/S1011134414001365]
- 4. Role of Hydrogen Bonding in Green Fluorescent Protein ... [https://www.nature.com/articles/s41598-019-47660-0]
- 5. Two Distinct Fluorescence States of the Ligand-Induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5770971/]
- 6. Discovery and Characterization of a Bilirubin Inducible ... [https://www.frontiersin.org/journals/marine-science/articles/10.3389/fmars.2021.678571/full]
- 7. Molecular engineering of UnaG: Insights into fluorescence ... [https://www.sciencedirect.com/science/article/abs/pii/S0168165625002160]
- 8. Imaging living obligate anaerobic bacteria with bilin- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7731933/]
- 9. Split Green Fluorescent Proteins: Scope, Limitations, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6537611/]
- 10. Genetically encoded fluorescent tags - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5385933/]
- 11. Fluorescent protein-based detection of unconjugated ... [https://www.nature.com/articles/srep28489]
- 12. Bilirubin as a Therapeutic Molecule: Challenges and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8532879/]
- 13. Green fluorescent protein [https://en.wikipedia.org/wiki/Green_fluorescent_protein]
- 14. The mechanism of cyclization in chromophore maturation ... [https://pubmed.ncbi.nlm.nih.gov/20593847/]
- 15. Direct observation of fluorescent proteins in gels [https://pmc.ncbi.nlm.nih.gov/articles/PMC11808229/]
- 16. Molecular dynamics guided identification of a brighter ... [https://www.nature.com/articles/s42004-025-01573-4]
- 17. Next-Generation Fluorogen-Based Reporters and ... [https://www.mdpi.com/1422-0067/20/24/6142]
- 18. Bright Near-Infrared Fluorescence Bio-Labeling With a ... [https://pubmed.ncbi.nlm.nih.gov/30471307/]
- 19. Enhanced small green fluorescent proteins as a ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.1039317/full]
- 20. 101: When Fluorescent lets you down Proteins GFP [https://blog.addgene.org/when-gfp-lets-you-down]
- 21. Imaging living obligate anaerobic bacteria with bilin- ... [https://www.sciencedirect.com/science/article/pii/S2666517420300018]
- 22. Live-cell imaging of cell signaling using genetically encoded ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5730511/]
- 23. New Orange Ligand-Dependent Fluorescent Reporter for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11180495/]
- 24. Live-cell imaging of circadian clock protein dynamics in ... [https://www.nature.com/articles/s41467-021-24086-9]
- 25. Next-Generation Fluorogen-Based Reporters and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6940837/]
- 26. Genetically encoded biosensors based on engineered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3000468/]
- 27. Adaptive Evolution of Eel Fluorescent Proteins from Fatty Acid ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0140972]
- 28. Eel green fluorescent protein is associated with resistance ... [https://pubmed.ncbi.nlm.nih.gov/26746389/]
- 29. Fluorescent proteins for a brighter science [https://www.sciencedirect.com/science/article/abs/pii/S0006291X22012281]
- 30. Fluorometric Nanoscale Analysis of Bilirubin and Biliverdin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272545/]
- 31. Fast, three-dimensional, live-cell super-resolution imaging ... [https://www.nature.com/articles/s41566-025-01638-9]
- 32. PET-based tracking of CAR T cells and viral gene transfer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12623248/]
- 33. Direct visualization of chimeric antigen receptors on ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1632823/full]
- 34. Nanoparticle-based Cell Trackers for Biomedical Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC6993224/]
- 35. Using Fluorescent Recombinant Proteins to Validate CAR- ... [https://www.aatbio.com/resources/application-notes/using-fluorescent-recombinant-proteins-to-validate-car-t-cell-therapy]
- 36. CAR Gene Delivery by T‐cell Targeted Lentiviral Vectors is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10724389/]
- 37. A convenient viral transduction based method for ... [https://www.sciencedirect.com/science/article/pii/S1687157X24001495]
- 38. Fluorescent proteins at a glance - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3037093/]
- 39. Experimental design of systems involving multiple ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250913004284]
- 40. Beyond the Green Fluorescent Protein: Biomolecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7033020/]
- 41. Bilirubin Binding - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/bilirubin-binding]
- 42. Fluorescent Proteins for Investigating Biological Events in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6032295/]
- 43. Identification and Characterisation of a pH-stable GFP [https://www.nature.com/articles/srep28166]
- 44. Bright and stable: New acid-tolerant green fluorescent ... [https://www.sciencedaily.com/releases/2018/01/180104120323.htm]
- 45. A Guide to Choosing Fluorescent Protein Combinations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8008483/]
- 46. Tips to Minimize Autofluorescence [https://fluorofinder.com/autofluorescence/]
- 47. Overcoming Autofluorescence to Assess GFP Expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6067662/]
- 48. Elimination of autofluorescence background from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3566262/]
- 49. Strong increase in the autofluorescence of cells signals ... [https://www.nature.com/articles/s41598-018-30623-2]
- 50. Combating Autofluorescence in Multiplex IF Image Analysis [https://oraclebio.com/overcoming-autofluorescence-variability-innovative-strategies-for-accurate-data/]
- 51. Systematic characterization of maturation time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5765880/]
- 52. Comparison among bright green fluorescent proteins in C. ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11787626/]
- 53. A Novel Fluorescence-Based Assay for the Measurement ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5785779/]
- 54. Bilirubin Targeting WNK1 to Alleviate NLRP3‐Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12362796/]
- 55. Comparative assessment of fluorescent proteins for in vivo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5221575/]
- 56. In vivo fluorescence imaging: success in preclinical imaging ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-022-01648-7]
- 57. The inducible chemical-genetic fluorescent marker FAST ... [https://www.nature.com/articles/s41598-018-28643-z]
- 58. Nanoscale Bilirubin Analysis in Translational Research and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10671013/]